Get your patient on Vonjo (Pacritinib)
Vonjo patient education
Patient toolkit
Dosage & administration
Coverage
See specific coverage requirements, including prior authorization and step therapies.

Vonjo prescribing information
Warnings and Precautions (5.9 ) 11/2024
INDICATIONS AND USAGE
VONJO is indicated for the treatment of adults with intermediate or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis (MF) with a platelet count below 50 × 10 9 /L.
This indication is approved under accelerated approval based on spleen volume reduction [see Clinical Studies (14 )] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
DOSAGE AND ADMINISTRATION
Recommended Dosage
The recommended dosage of VONJO is 200 mg orally twice daily. VONJO may be taken with or without food.
Swallow capsules whole. Do not open, break, or chew capsules.
Patients who are on treatment with other kinase inhibitors before the initiation of VONJO must taper or discontinue according to the prescribing information for that drug.
Monitoring for Safety
Perform a complete blood count (CBC; including white blood cell count differential and platelet count), coagulation testing (prothrombin time, partial thromboplastin time, thrombin time, and international normalized ratio) and a baseline electrocardiogram (ECG), prior to starting VONJO, and monitor as clinically indicated while the patient is on treatment.
Missed Dose
If a dose of VONJO is missed, the patient should take the next prescribed dose at its scheduled time. Extra capsules should not be taken to make up for the missed dose.
Dose Interruption for Planned Surgical Procedures or Other Interventions
Discontinue VONJO 7 days prior to elective surgery or invasive procedures because of the risk of hemorrhage and restart only after hemostasis is assured.
Dose Modification for Adverse Reactions
Dose modifications for diarrhea, thrombocytopenia, hemorrhage, and prolonged QT interval are described in Table 1 , Table 2 , Table 3 , and Table 4 respectively. See Warning and Precautions (5.1 , 5.2 , 5.3 , and 5.4 ) for additional risk minimization recommendations.
Dose levels for VONJO are as follows: 200 mg twice daily (initial starting dose), 100 mg twice daily (first dose reduction), 100 mg once daily (second dose reduction). Discontinue VONJO in patients unable to tolerate a dose of 100 mg daily.
Table 1 Dosage Modification for Diarrhea
| Toxicity | Management/Action |
|---|---|
| a Increase of at least 7 stools per day over baseline, or hospitalization indicated, or severe increase in ostomy output over baseline, or if limiting self-care. | |
| b Increase of <4 stools per day over baseline or mild increase in ostomy output compared to baseline. | |
| New onset of diarrhea |
|
| Grade 3 or 4 a |
|
Table 2 Dose Modification for Thrombocytopenia
| Worsening Thrombocytopenia | Action |
|---|---|
| For clinically significant worsening of thrombocytopenia that lasts more than 7 days |
|
Table 3 Dose Modification for Hemorrhage
| Toxicity | Action |
|---|---|
| Moderate bleeding; intervention indicated |
|
| Severe bleeding; transfusion, invasive intervention, or hospitalization indicated |
|
| Life-threatening bleeding; urgent intervention indicated. |
|
Table 4 Dose Modification for Prolonged QT Interval
| Toxicity | Action |
|---|---|
| QTc prolongation >500 msec or >60 msec from baseline |
|
Dosage Modification for Patients with Severe Hepatic Impairment
The recommended dosage of VONJO in patients with severe hepatic impairment [Child-Pugh C] is 100 mg twice daily. Dosage may be increased to 200 mg twice daily if the treatment is not effective after 12 weeks and there are no safety concerns; continue monitoring for safety [see Use in Specific Populations (8.6 ) and Clinical Pharmacology (12.3 )].
DOSAGE FORMS AND STRENGTHS
Capsule: 100 mg, oblong, size 0 hard gelatin capsule with an opaque scarlet cap printed with “Pacritinib 100 mg” and opaque gray body printed with “C78837”.
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed (8.2 ).
Hepatic Impairment: For patients with severe hepatic impairment (Child-Pugh C), the recommended dosage is 100 mg twice daily (8.6 ).
Renal Impairment: Avoid use in patients with eGFR <30 mL/min (8.7 ).
Pregnancy
Risk Summary
There are no available data on VONJO use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, administration of pacritinib to pregnant mice or rabbits at exposures that were considerably lower than those observed at the recommended human dose were associated with maternal toxicity and embryonic and fetal loss (see Data ) . Advise pregnant women of the potential risk to a fetus. Consider the benefits and risks of VONJO for the mother and possible risks to the fetus when prescribing VONJO to a pregnant woman.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Pacritinib was administered orally to pregnant mice at doses of 30, 100, or 250 mg/kg/day from gestation day 6 to gestation day 15. Pacritinib was also administered orally to pregnant rabbits at doses of 15, 30, or 60 mg/kg/day from gestation day 7 until gestation day 20. In both species, pacritinib was associated with maternal toxicity, which resulted in post-implantation loss in mice, abortions in rabbits, and reduced fetal body weights in mice and rabbits at exposures 0.1 times (mice) and 0.3 times (rabbits) the exposure at the recommended human dose (AUC-based). In mice, the high dose was associated with an increased incidence of an external malformation (cleft palate) in the presence of maternal toxicity.
In a pre- and post-natal development study in mice, pregnant animals were dosed with pacritinib from implantation through lactation at 30, 100, or 250 mg/kg/day. Maternal toxicity was noted at 250 mg/kg and associated with increased gestation length and dystocia, lowered mean birth weights and neonatal survival, and transiently delayed startle response, learning, and memory development at weaning.
Lactation
Risk Summary
There are no data on the presence of pacritinib in either human or animal milk, the effects on the breastfed child, or the effects on milk production. It is not known whether VONJO is excreted in human milk. Because of the potential for serious adverse reactions in the breastfed child, advise patients that breastfeeding is not recommended during treatment with VONJO, and for 2 weeks after the last dose.
Females and Males of Reproductive Potential
Infertility
Males
Pacritinib reduced male mating and fertility indices in BALB/c mice [see Nonclinical Toxicology (13.1 )]. Pacritinib may impair male fertility in humans.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Clinical studies of VONJO did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects.
Hepatic Impairment
In patients with severe hepatic impairment [Child-Pugh C], the recommended dosage of VONJO is 100 mg twice daily. Dosage may be increased to 200 mg twice daily if the treatment is not effective after 12 weeks and there are no safety concerns. Monitor hepatically-impaired patients for adverse reactions and consider VONJO dose modifications based on safety and efficacy [see Dosage and Administration (2.5), Clinical Pharmacology (12.3 )].
No dose modification is recommended for patients with mild [Child-Pugh A] or moderate [Child-Pugh B] hepatic impairment [see Clinical Pharmacology (12.3 )].
Renal Impairment
Administration of a single dose of VONJO 400 mg to subjects with renal impairment resulted in approximately 30% increase in C max and AUC of pacritinib in subjects with eGFR 15 to 29 mL/min and eGFR <15 mL/min on hemodialysis compared to subjects with normal renal function (eGFR ≥90 mL/min). Avoid use of VONJO in patients with eGFR <30 mL/min [see Clinical Pharmacology (12.3 )] .
CONTRAINDICATIONS
VONJO is contraindicated in patients concomitantly using strong CYP3A4 inhibitors or inducers as these medications can significantly alter exposure to pacritinib, which may increase the risk of adverse reactions or impair efficacy [see Warnings and Precautions (5.1 , 5.2 , 5.3 , 5.4 ), Drug Interactions (7.1 ), and Clinical Pharmacology (12.3 )] .
WARNINGS AND PRECAUTIONS
- Hemorrhage: Avoid use in patients with active bleeding and hold VONJO prior to any planned surgical procedures. May require dose interruption, dose reduction or permanent discontinuation depending on severity (5.1 ).
- Diarrhea: Manage significant diarrhea with anti-diarrheals, dose reduction, or dose interruption (5.2 ).
- Thrombocytopenia: Manage by dose reduction or interruption (5.3 ).
- Prolonged QT Interval: Avoid use in patients with baseline QTc >480 msec. Interrupt and reduce VONJO dosage in patients who have a QTcF >500 msec. Correct hypokalemia prior to and during VONJO administration (5.4 ).
- Major Adverse Cardiac Events (MACE): Risk may be increased in current/past smokers and patients with other cardiovascular risk factors. Monitor for signs, evaluate and treat promptly (5.5 ).
- Thrombosis: Including deep venous thrombosis, pulmonary embolism, and arterial thrombosis may occur. Monitor for signs, evaluate and treat promptly (5.6 ).
- Secondary Malignancies: Lymphoma and other malignancies may occur. Past/current smokers may be at increased risk (5.7 ).
- Risk of Infection: Delay starting VONJO until active serious infections have resolved. Observe for signs and symptoms of infection and manage promptly (5.8 ).
- Symptom Exacerbation Following Interruption or Discontinuation: Manage with supportive care and consider resuming treatment with VONJO (5.9 ).
Hemorrhage
Serious (11%) and fatal (2%) hemorrhages have occurred in VONJO-treated patients with platelet counts <100 x 10 9 /L. Serious (13%) and fatal (2%) hemorrhages have occurred in VONJO-treated patients with platelet counts <50 x 10 9 /L. Grade ≥3 bleeding events (defined as requiring transfusion or invasive intervention) occurred in 15% of patients treated with VONJO compared to 7% of patients treated on the control arm. Due to hemorrhage, VONJO dose-reductions, dose interruptions, or permanent discontinuations occurred in 3%, 3%, and 5% of patients, respectively.
Avoid use of VONJO in patients with active bleeding and hold VONJO 7 days prior to any planned surgical or invasive procedures.
Assess platelet counts periodically, as clinically indicated [see Warnings and Precautions (5.3 )] . Manage hemorrhage using treatment interruption and medical intervention [see Dosage and Administration (2.5 )] .
Diarrhea
VONJO caused diarrhea in approximately 48% of patients compared to 15% of patients treated on the control arm in a clinical trial. The median time to resolution in VONJO-treated patients was 2 weeks. The incidence of reported diarrhea decreased over time with 41% of patients reporting diarrhea in the first 8 weeks of treatment, 15% in Weeks 8 through 16, and 8% in Weeks 16 through 24. Diarrhea resulted in treatment interruption in 3% of VONJO-treated patients. Serious diarrhea adverse reactions occurred in 2% of patients treated with VONJO compared to no such adverse reactions in patients in the control arm. In postmarketing reports, severe diarrhea leading to acute kidney injury and treatment discontinuation has been reported with VONJO.
Control pre-existing diarrhea before starting VONJO treatment. Manage diarrhea with antidiarrheal medications, fluid replacement, and dose-modification. Upon initiation of therapy, prescribe an anti-diarrheal medication (e.g., loperamide), and instruct patients to treat diarrhea promptly at the first onset of symptoms (change in frequency or consistency of bowel movements) after starting VONJO. Interrupt or reduce VONJO dose in patients with significant diarrhea despite optimal supportive care [see Dosage and Administration (2.5 )] .
Thrombocytopenia
VONJO can cause worsening thrombocytopenia. VONJO dosing was reduced due to worsening thrombocytopenia in 2% of patients with pre-existing moderate to severe thrombocytopenia (platelet count <100 x 10 9 /L). VONJO dosing was reduced due to worsening thrombocytopenia in 2% of patients with pre-existing severe thrombocytopenia (platelet count <50 x 10 9 /L).
Monitor platelet count prior to VONJO treatment and as clinically indicated during treatment [see Dosage and Administration (2.2 )] . Interrupt VONJO in patients with clinically significant worsening of thrombocytopenia that lasts for more than 7 days. Restart VONJO at 50% of the last given dose once the toxicity has resolved. If toxicity recurs hold VONJO. Restart VONJO at 50% of the last given dose once the toxicity has resolved [see Dosage and Administration (2.5 )] .
Prolonged QT Interval
VONJO can cause prolongation of the QTc interval. QTc prolongation of >500 msec was higher in VONJO-treated patients than in patients in the control arm (1.4% vs 1%). QTc increase from baseline by 60 msec or higher was greater in VONJO-treated patients than in control arm patients (1.9% vs 1%). Adverse reactions of QTc prolongation were reported for 3.8% of VONJO-treated patients and 2% of control arm patients. No cases of torsades de pointes were reported.
Avoid use of VONJO in patients with a baseline QTc of >480 msec. Avoid use of drugs with significant potential for QTc prolongation in combination with VONJO. Correct hypokalemia prior to and during VONJO treatment.
Manage QTc prolongation using VONJO interruption and electrolyte management [see Dosage and Administration (2.5 )] .
Major Adverse Cardiac Events (MACE)
Another Janus associated kinase (JAK)-inhibitor has increased the risk of MACE, including cardiovascular death, myocardial infarction, and stroke (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which VONJO is not indicated.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with VONJO particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Another JAK-inhibitor has increased the risk of thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which VONJO is not indicated.
Patients with symptoms of thrombosis should be promptly evaluated and treated appropriately.
Secondary Malignancies
Another JAK-inhibitor has increased the risk of lymphoma and other malignancies excluding non-melanoma skin cancer (NMSC) (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which VONJO is not indicated. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with VONJO, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy, and patients who are current or past smokers.
Risk of Infection
Another JAK-inhibitor increased the risk of serious infections (compared to best available therapy) in patients with myeloproliferative neoplasms. Serious bacterial, mycobacterial, fungal and viral infections may occur in patients treated with VONJO. Delay starting therapy with VONJO until active serious infections have resolved. Observe patients receiving VONJO for signs and symptoms of infection and manage promptly. Use active surveillance and prophylactic antibiotics according to clinical guidelines.
Symptom Exacerbation Following Interruption or Discontinuation of Treatment
Following discontinuation of JAK-inhibitors, including VONJO, signs and symptoms from myeloproliferative neoplasms may flare. Some patients with MF have experienced one or more of the following after discontinuing JAK-inhibitors: fever, respiratory distress, hypotension, disseminated intravascular coagulation, or multi-organ failure.
If one or more of these signs and symptoms occur after discontinuation of VONJO, evaluate for and treat any intercurrent illness and consider restarting VONJO. Instruct patients not to interrupt or discontinue therapy without consulting their healthcare provider. When discontinuing or interrupting therapy with VONJO for reasons other than potentially life-threatening toxicities, consider tapering the dose of VONJO gradually rather than discontinuing abruptly [see Dosage and Administration (2.5 )] .
Interactions With CYP3A4 Inhibitors or Inducers
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hemorrhage [see Warnings and Precautions (5.1 )]
- Diarrhea [see Warnings and Precautions (5.2 )]
- Thrombocytopenia [see Warnings and Precautions (5.3 )]
- Prolonged QT Interval [see Warnings and Precautions (5.4 )]
- Major Adverse Cardiac Events [see Warnings and Precautions (5.5 )]
- Thrombosis [see Warnings and Precautions (5.6 )]
- Secondary Malignancies [see Warnings and Precautions (5.7 )]
- Risk of Infection [see Warnings and Precautions (5.8 )]
- Symptom Exacerbation Following Interruption or Discontinuation of Treatment [see Warnings and Precautions (5.9 )]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
PERSIST-2 Trial
The safety of VONJO was evaluated in the randomized, controlled PERSIST-2 trial [see Clinical Studies (14 )] . In PERSIST-2, key eligibility criteria included adults with intermediate or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) MF with splenomegaly and a platelet count ≤100 × 10 9 /L. Prior Janus associated kinase (JAK) inhibitor therapy was permitted. Patients received VONJO at 200 mg twice daily (n=106), 400 mg once daily (n=104), or best available therapy (BAT) (n=98). Forty-seven (44%) of the 106 patients treated with VONJO 200 mg twice daily had a baseline platelet count of <50 × 10 9 /L. The 400 mg once daily dose could not be established to be safe, so further information on this arm is not provided.
In PERSIST-2, among the 106 patients treated with VONJO 200 mg twice daily, the median baseline hemoglobin was 9.7 g/dL and the median drug exposure was 25 weeks. Fifty-four percent of patients were exposed for 6 months, and 18% were exposed for approximately 12 months. Accounting for dose reductions, the average daily dose (mean relative dose intensity) and median daily dose (median relative dose intensity) were 380 mg (95%) and 400 mg (100%), respectively, for patients receiving VONJO twice daily.
The median age of patients who received VONJO 200 mg twice daily was 67 years (range: 39 to 85 years), 59% were male, 86% were White, 3% were Asian, 2% were Native Hawaiian or Other Pacific Islander, 0% were Black, 9% did not report race, and 87% had an Eastern Cooperative Oncology Group performance status of 0 to 1.
Serious adverse reactions occurred in 47% of patients treated with VONJO 200 mg twice daily and in 31% of patients treated with BAT. The most frequent serious adverse reactions occurring in ≥3% patients receiving VONJO 200 mg twice daily were anemia (8%), thrombocytopenia (6%), pneumonia (6%), cardiac failure (4%), disease progression (3%), pyrexia (3%), and squamous cell carcinoma of skin (3%). Fatal adverse reactions occurred in 8% of patients receiving VONJO 200 mg twice daily and in 9% of patients treated with BAT. The fatal adverse reactions among patients treated with VONJO 200 mg twice daily included events of disease progression (3%), and multiorgan failure, cerebral hemorrhage, meningorrhagia, and acute myeloid leukemia in <1% of patients each, respectively.
Permanent discontinuation due to an adverse reaction occurred in 15% of patients receiving VONJO 200 mg twice daily compared to 12% of patients treated with BAT. The most frequent reasons for permanent discontinuation in ≥2% of patients receiving VONJO 200 mg twice daily included anemia (3%) and thrombocytopenia (2%).
Drug interruptions due to an adverse reaction occurred in 27% of patients who received VONJO 200 mg twice daily compared to 10% of patients treated with BAT. The most frequent reasons for drug interruption in ≥2% of patients receiving VONJO 200 mg twice daily were anemia (5%), thrombocytopenia (4%), diarrhea (3%), nausea (3%), cardiac failure (3%), neutropenia (2%), and pneumonia (2%).
Dosage reductions due to an adverse reaction occurred in 12% of patients who received VONJO 200 mg twice daily compared to 7% of patients treated with BAT. Adverse reactions requiring dosage reduction in ≥2% of patients who received VONJO 200 mg twice daily included thrombocytopenia (2%), neutropenia (2%), conjunctival hemorrhage (2%), and epistaxis (2%).
The most common adverse reactions in ≥20% of patients (N=106) were diarrhea, thrombocytopenia, nausea, anemia, and peripheral edema.
Table 5 summarizes the common adverse reactions in PERSIST-2 during randomized treatment.
Table 5 Adverse Reactions Reported in ≥10% Patients Receiving VONJO 200 mg Twice Daily or Best Available Therapy During Randomized Treatment in PERSIST-2
| a Grade by CTCAE Version 4.03 | ||||
| Adverse Reactions | VONJO (200 mg Twice Daily) (N=106) | Best Available Therapy (N=98) | ||
| All Grades a % | Grade ≥3 % | All Grades a % | Grade ≥3 % | |
| Diarrhea | 48 | 4 | 15 | 0 |
| Thrombocytopenia | 34 | 32 | 23 | 18 |
| Nausea | 32 | 1 | 11 | 1 |
| Anemia | 24 | 22 | 15 | 14 |
| Peripheral edema | 20 | 1 | 15 | 0 |
| Vomiting | 19 | 0 | 5 | 1 |
| Dizziness | 15 | 1 | 5 | 0 |
| Pyrexia | 15 | 1 | 3 | 0 |
| Epistaxis | 12 | 5 | 13 | 1 |
| Dyspnea | 10 | 0 | 9 | 3 |
| Pruritus | 10 | 2 | 6 | 0 |
| Upper respiratory tract infection | 10 | 0 | 6 | 0 |
| Cough | 8 | 2 | 10 | 0 |
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VONJO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- Renal: Acute or subacute kidney injury (secondary to diarrhea) [see Warnings and Precautions (5.2)]
DRUG INTERACTIONS
Co-administration of VONJO with moderate CYP3A4 inhibitors can increase the exposure to pacritinib. Monitor for increased adverse reactions of VONJO when administered with moderate CYP3A4 inhibitors (7.1 ).
VONJO is an inhibitor of P-gp, BCRP, and CYP1A2 and an inducer of CYP3A4 and CYP2C19. Monitor patients concomitantly receiving substrates of these transporters and enzymes, and adjust dose of the substrates as needed (7.2 ).
VONJO may reduce the effectiveness of hormonal contraceptives (7.2 )
Effect of Other Drugs on VONJO
Strong and Moderate CYP3A4 Inhibitors
Pacritinib is predominantly metabolized by CYP3A4. Concomitant use of VONJO with strong and moderate CYP3A4 inhibitors increases pacritinib exposure, which may increase the risk of exposure-related adverse reactions [see Clinical Pharmacology (12.3 )]
Co-administration of VONJO with strong CYP3A4 inhibitors is contraindicated [see Contraindications (4 )].
Monitor patients concomitantly receiving moderate CYP3A4 inhibitors (e.g., fluconazole) for increased adverse reactions and consider VONJO dose modifications based on safety [see Dosage and Administration (2.5 )] . Concomitant use of VONJO with doses of fluconazole greater than 200 mg once daily has not been studied.
Strong CYP3A4 Inducers
Pacritinib is predominantly metabolized by CYP3A4. Concomitant use of VONJO with strong CYP3A4 inducers decreases pacritinib exposure, which may reduce efficacy of VONJO [see Clinical Pharmacology (12.3 )] .
Co-administration of VONJO with strong CYP3A4 inducers is contraindicated [see Contraindications (4 )].
Effect of VONJO on Other Drugs
CYP1A2 Substrates
Pacritinib is an inhibitor of CYP1A2. VONJO increases the plasma concentrations of CYP1A2 substrates [see Clinical Pharmacology (12.3 )] ,which may increase the risk of adverse reactions from the CYP1A2 substrate.
Monitor for CYP1A2 substrate related adverse reactions more frequently, unless otherwise recommended in the substrate Prescribing Information, when VONJO is used concomitantly with CYP1A2 substrates where minimal substrate concentration changes may lead to serious adverse reactions.
CYP2C19 Substrates
Pacritinib is an inducer of CYP2C19. VONJO decreases the plasma concentrations of CYP2C19 substrates [see Clinical Pharmacology (12.3 )] , which may decrease the efficacy from the CYP2C19 substrate.
Monitor the efficacy of CYP2C19 substrate more frequently, unless otherwise recommended in the substrate Prescribing Information, when VONJO is used concomitantly with CYP2C19 substrates where minimal substrate concentration changes may lead to diminished efficacy. Dose adjustment of CYP2C19 substrates may be needed.
CYP3A4 Substrates
Pacritinib is an inducer of CYP3A4. VONJO decreases the plasma concentrations of CYP3A4 substrates [see Clinical Pharmacology (12.3 )] , which may decrease the efficacy from the CYP3A4 substrate.
Monitor the efficacy of CYP3A4 substrate more frequently, unless otherwise recommended in the substrate Prescribing Information, when VONJO is used concomitantly with CYP3A4 substrates where minimal substrate concentration changes may lead to diminished efficacy. Dose adjustment of CYP3A4 substrates may be needed.
Hormonal Contraceptives
Avoid concomitant use of VONJO with hormonal contraceptives except for intrauterine systems containing levonorgestrel. The effectiveness of hormonal contraceptives, except for intrauterine systems containing levonorgestrel, may be reduced when used with VONJO.
If contraception is needed or desired, an alternate contraceptive that is not affected by CYP3A4 inducers (e.g., an intrauterine system) or additional nonhormonal contraceptive (e.g., condoms) should be used when treated concomitantly with VONJO, and for 30 days after last dose of VONJO.
P-gp Substrates
Pacritinib is an inhibitor of P-gp. VONJO increases the plasma concentrations of P-gp substrates [see Clinical Pharmacology (12.3 )] , which may increase the risk of adverse reactions from the P-gp substrate.
Monitor for P-gp substrate related adverse reactions more frequently, unless otherwise recommended in the substrate Prescribing Information, when VONJO is used concomitantly with P-gp substrates where minimal substrate concentration changes may lead to serious adverse reactions.
Digoxin: Measure serum digoxin concentrations before initiating concomitant use with VONJO and continue monitoring serum digoxin concentrations as recommended in the Prescribing Information for digoxin [see Clinical Pharmacology (12.3 )] .
BCRP substrates
Pacritinib is an inhibitor of BCRP. VONJO increases the plasma concentrations of BCRP substrates [see Clinical Pharmacology (12.3 )] , which may increase the risk of adverse reactions from the BCRP substrate.
When used concomitantly with VONJO, monitor for BCRP substrate related adverse reactions more frequently and consider dose reduction of the BCRP substrate according to its Prescribing Information.
Rosuvastatin: The dose of rosuvastatin should not exceed 20 mg once daily when concomitantly used with VONJO [see Clinical Pharmacology (12.3 )] .
DESCRIPTION
VONJO contains pacritinib citrate, a kinase inhibitor with the chemical name (2E,16E)-11-[2-(pyrrolidin-1-yl)ethoxy]-14,19-dioxa-5,7,27-triazatetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2,4,6,8,10,12(26),16,21,23-decaene citrate and a molecular weight of 664.7 as citrate salt and 472.59 as a free base. The molecular formula is C 28 H 32 N 4 O 3 •C 6 H 8 O 7 and the structural formula is:
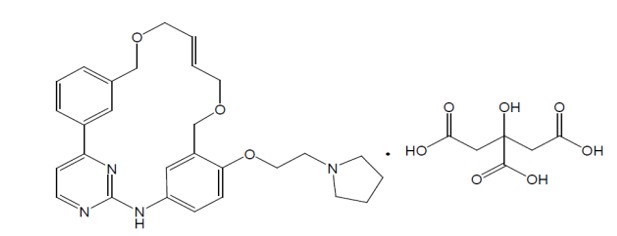
VONJO capsule is for oral administration. Each capsule contains 100 mg of pacritinib equivalent to 140.65 mg of pacritinib citrate and the inactive ingredients are microcrystalline cellulose NF, polyethylene glycol 8000 (PEG 8000) NF, and magnesium stearate NF. The gelatin capsule is bovine derived. The capsule shell contains gelatin, titanium dioxide, black iron oxide, erythrosine, red iron oxide, and printing ink containing shellac, propylene glycol, titanium dioxide, sodium hydroxide, and povidone.
CLINICAL PHARMACOLOGY
Mechanism of Action
Pacritinib is an oral kinase inhibitor with activity against wild type Janus associated kinase 2 (JAK2), mutant JAK2V617F, FMS-like tyrosine kinase 3 (FLT3), and interleukin 1 receptor associated kinase-1 (IRAK1) which contribute to signaling of a number of cytokines and growth factors that are important for hematopoiesis and immune function. Pacritinib is also an inhibitor of activin A receptor, type 1/activin receptor like-kinase 2 (ACVR1/ALK2).
MF is often associated with dysregulated JAK2 signaling. At clinically relevant concentrations, pacritinib does not inhibit JAK1. Pacritinib has higher inhibitory activity for JAK2 compared to JAK3 and tyrosine kinase 2 (TYK2). Pacritinib exhibits inhibitory activity against additional cellular kinases, such as colony stimulating factor 1 receptor (CSF1R), of which the clinical relevance in myelofibrosis is unknown.
Pharmacodynamics
Pacritinib inhibited the phosphorylation of signal transducer and activator of transcription 5 (STAT5) protein in a dose-dependent manner (ex vivo) in expanded erythroid progenitor cells derived from healthy subjects. Administration of single doses of 400 mg pacritinib resulted in modest inhibition of interleukin-6-induced STAT3 phosphorylation in whole blood derived from healthy subjects.
Cardiac Electrophysiology
In a 24-week study of 54 patients with MF treated with VONJO 200 mg twice daily, the maximum mean (90% confidence interval) change in QTcF from baseline was 11 (90% CI: 5-17) msec.
Pharmacokinetics
Pacritinib steady-state mean (CV%) C max is 8.4 mg/L (32.4%) and AUC 0-12 is 95.6 mg×h/L (33.1%) following administration of VONJO 200 mg twice daily in patients with MF. Pharmacokinetics of pacritinib increases in a less than dose-proportional manner. VONJO 200 mg twice daily accumulates 386% and reaches steady-state within a week.
Absorption
Pacritinib achieves C max within approximately 4 to 5 hours post-dose.
Effect of Food
There was no significant effect of food on the pharmacokinetics of pacritinib following oral administration of VONJO 200 mg with a high-fat meal.
Distribution
The median (range) apparent volume of distribution of pacritinib at steady state is 229 L (156 to 591 L) in patients with MF taking 200 mg twice daily. Plasma protein binding of pacritinib is approximately 98.8%.
Metabolism
Pacritinib is predominantly metabolized by the CYP3A4 isozyme. Pacritinib is the major circulating component and the pharmacologic activity is mainly attributed to the parent molecule. Two major metabolites, M1 and M2, in human whole plasma represent 9.6% and 10.5% of parent drug exposure, respectively.
Elimination
The mean apparent clearance at steady-state (CV%) of pacritinib is 2.09 L/h (33.1%), and mean effective half-life (CV%) is 27.7 hours (17.0%).
Excretion
Following a single oral administration of radiolabeled pacritinib 400 mg in healthy adult subjects, 87% of the radioactivity was recovered in feces, and 6% was recovered in urine. No unchanged drug was excreted in feces and 0.12% of unchanged drug was excreted in urine.
Specific Populations
No clinically significant differences in the pharmacokinetics of pacritinib were observed based on age, sex, body weight, or race.
Patients With Renal Impairment
Pacritinib C max and AUC were similar in subjects with eGFR 30 to 89 mL/min, as estimated by the MDRD study equation, compared to subjects with eGFR ≥90 mL/min. The C max and AUC increased approximately 30% in subjects with eGFR 15 to 29 mL/min and eGFR <15 mL/min on hemodialysis.
Patients With Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of pacritinib was studied in subjects with normal or impaired hepatic function at 200 mg VONJO twice daily for 14 days. The geometric mean AUC of total pacritinib decreased in subjects with moderate [Child-Pugh B, n=8] or severe hepatic impairment [Child-Pugh C, n=6] compared to subjects with normal hepatic function (n=11) due to reduced plasma protein binding. The geometric mean AUC of unbound pacritinib, the pharmacologically relevant parameter, was similar in healthy subjects compared to subjects with moderate hepatic impairment and increased by 23% in subjects with severe hepatic impairment (see Table 6).
Table 6 Steady-State Pharmacokinetic Changes in Pacritinib at 200 mg VONJO BID: Subjects with Moderate / Severe Hepatic Impairment vs. Normal Hepatic Function
Hepatic impairment | Ratio (90% CI) of pacritinib exposure 1 | |
Unbound C max | Unbound AUC 0-tau | |
Moderate impairment (Child-Pugh B) | 1.06 (0.72, 1.58) | 1.05 (0.69, 1.60) |
Severe impairment (Child-Pugh C) | 1.24 (0.93, 1.65) | 1.23 (0.91, 1.66) |
Notes: AUC 0-tau = Area under the curve from zero to tau post-dose after 14 days twice daily dosing with VONJO 200 mg; CI = Confidence Interval; C max = Maximum plasma concentration.
1 Ratios for C max and AUC compare exposure of unbound pacritinib in subjects with hepatic impairment vs normal hepatic function following administration of VONJO 200 mg BID for 14 days.
Drug Interactions
Effects of Other Drugs on the Pharmacokinetics of VONJO
The effect of co-administered drugs on the exposure of pacritinib is shown in Table 7 and Table 8.
Table 7 Change in Pharmacokinetics of Pacritinib Following Administration of a Single 400 mg Dose VONJO in the Presence of Co-administered Drugs
Co-administered Drug | Regimen of Co-administered Drug | Ratio (90% CI) of pacritinib exposure 1 | |
C max | AUC 0-t | ||
Clarithromycin (strong CYP3A4 inhibitor) | 500 mg every 12 hours for 5 days | 1.30 (1.22, 1.39) | 1.80 (1.67, 1.94) |
Rifampin (strong CYP3A4 inducer) | 600 mg once daily for 10 days | 0.49 (0.43, 0.55) | 0.13 (0.11, 0.15) |
Notes: AUC 0-t = Area under the curve from zero to the last quantifiable concentration; CI = Confidence interval; CYP = Cytochrome P450 1 Ratios for C max and AUC compare co-administration of the medication with VONJO vs. administration of single dose 400 mg VONJO alone
Table 8 Change in Pharmacokinetics of 200 mg VONJO BID at Steady State in the Presence of Co-administered Drugs
Co-administered Drug | Regimen of Co-administered Drug | Ratio (90% CI) of pacritinib exposure 1 | |
C max | AUC 0-tau | ||
Fluconazole (moderate CYP3A4 inhibitor) | 200 mg once daily for 7 days | 1.41 (1.35, 1.48) | 1.45 (1.39, 1.52) |
Bosentan (moderate CYP3A4 inducer) | 125 mg twice daily for 7 days | 0.84 (0.76, 0.92) | 0.79 (0.72, 0.87) |
Notes: AUC 0-tau = Area under the curve for a dosing interval; BID = Twice daily; CI = Confidence interval; C max = Maximum plasma concentration; CYP = Cytochrome P450. 1 Ratios for C max and AUC 0-tau compare co-administration of the medication with 200 mg VONJO BID vs. administration of 200 mg VONJO alone at steady state.
Effects of VONJO on the Pharmacokinetics of Other Drugs
The effect of pacritinib on the exposure of other co-administered drugs is shown in Table 9.
Table 9 Change in Pharmacokinetics of Co-administered Drugs with 200 mg VONJO BID at Steady State
Co-administered Drug | Regimen of Co-administered Drug (single dose) | Ratio (90% CI) of exposure of co-administered drug 1 | |
C max | AUC | ||
Caffeine (CYP1A2 substrate) | 100 mg | 0.99 (0.92, 1.07) | 1.22 (1.12, 1.32) |
Midazolam (CYP3A4 substrate) | 2 mg | 0.40 (0.34, 0.46) | 0.40 (0.35, 0.46) |
Omeprazole (CYP2C19 substrate) | 20 mg | 0.73 (0.46, 1.15) | 0.49 (0.27, 0.89) |
Notes: AUC = Area under the curve from zero to the last quantifiable concentration displayed for caffeine and AUC from zero extrapolated to infinity displayed for midazolam and omeprazole; BID = Twice daily; CI = Confidence interval; C max = Maximum plasma concentration; CYP = Cytochrome P450.
1 Ratios for C max and AUC compare a single dose of co-administered drug with 200 mg VONJO BID at steady state vs. administration of a single dose of the co-administered drug alone.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Pacritinib is a time-dependent inhibitor of CYP1A2 and CYP3A4, and a reversible inhibitor of CYP3A4 and CYP2C19 (Ki ≤10 µM). Pacritinib shows less direct inhibition towards CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP2D6 (Ki >10 µM). Pacritinib is an inducer of CYP1A2 and CYP3A4.
Transporter Systems
Table 10 Change in Pharmacokinetics of Co-administered Drugs with 200 mg VONJO BID at Steady State
Co-administered Drug | Regimen of Co-administered Drug (single dose) | Ratio (90% CI) of exposure of co-administered drug 1 | |
C max | AUC | ||
Metformin (OCT1 substrate) | 500 mg | 0.96 (0.87, 1.07) | 1.05 (0.96, 1.15) |
Digoxin (P-gp substrate) | 0.25 mg | 1.29 (1.13, 1.47) | 1.15 (1.05, 1.27) |
Rosuvastatin (BCRP substrate) | 5 mg | 2.04 (1.79, 2.31) | 1.80 (1.63, 1.99) |
Notes: AUC = Area under the curve from zero to the last quantifiable concentration displayed for digoxin and rosuvastatin and AUC from zero extrapolated to infinity displayed for metformin; BID = Twice daily; CI = Confidence interval; C max = Maximum plasma concentration; CYP = Cytochrome P450
1 Ratios for C max and AUC compare a single dose of co-administered drug with 200 mg VONJO BID at steady state vs. administration of a single dose of the co-administered drug alone.
In Vitro Studies
Transporter Systems: Pacritinib is not a substrate of BCRP, MRP2, OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, or P-gp. Pacritinib is an inhibitor of BCRP, OCT1, OCT2, and P-gp. Pacritinib is not an inhibitor of BSEP, MRP2, OAT1, or OAT3.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Pacritinib was not carcinogenic in the 6-month Tg.rasH2 transgenic mouse model. Pacritinib was not carcinogenic in a 2-year carcinogenicity study in rats at 0.004 times and 0.014 times, in males and females, respectively, the recommended human dose (AUC-based). Pacritinib exposures achieved in mice and rats during the carcinogenicity assessments were considerably lower than the exposure observed at the recommended human dose.
Pacritinib was not mutagenic in a bacterial mutagenicity assay (Ames test) or clastogenic in vitro in a chromosomal aberration assay (Chinese hamster ovary cells) or in vivo in a micronucleus test in mice.
In a fertility study in male BALB/c mice, pacritinib was administered for at least 70 days prior to cohabitation with untreated partners. Pacritinib had no effects at any dose level on uterine implantation, macroscopic findings, reproductive organ weights, and sperm evaluations. At 213.4 mg/kg/day (3.0 times, the recommended human dose, based on body surface area), reduced mating and fertility indices were observed in male BALB/c mice. In a fertility and early embryonic development study in CD-1 mice, no effects on male or female reproductive performance, including assessments of mating, fertility, estrous cyclicity, and intrauterine survival, were observed at doses up to 250 mg/kg/day (3.0 times, the recommended human dose, based on body surface area).
CLINICAL STUDIES
PERSIST-2
The efficacy of VONJO in the treatment of patients with intermediate or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) MF was established in the PERSIST-2 trial.
PERSIST-2 enrolled patients with intermediate or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) MF with splenomegaly and a platelet count ≤100 × 10 9 /L. Both JAK2 naïve patients and patients with prior JAK2 inhibitor therapy were included. Patients were randomized 1:1:1 to receive VONJO 400 mg once daily, VONJO 200 mg twice daily, or best available therapy (BAT). BAT agents could be used alone, in combinations, sequentially, and intermittently, as clinically indicated by standards of care. BAT included any physician-selected treatment for MF and may have included ruxolitinib, hydroxyurea, glucocorticoids, erythropoietic agents, immunomodulatory agents, mercaptopurine, danazol, interferons, cytarabine, melphalan. BAT also included no treatment (“watch and wait”) or symptom-directed treatment without MF-specific treatment.
In this trial, 311 patients were randomized to receive VONJO 400 mg once daily (n=104), VONJO 200 mg twice daily (n=107), or BAT (n=100). The VONJO dose of 400 mg once daily was not established as safe and is not an approved dosage regimen.
The demographic characteristics of the efficacy population were median age of 68 years (range 32 to 91), 55% male, 86% Caucasian, and 14% non-Caucasian. The VONJO and BAT treatment arms were well balanced with respect to age, gender, race, ethnicity, body mass index, and geographic region. Sixty-eight percent of patients had primary MF, 20% had post-polycythemia vera MF, and 12% had post-essential thrombocythemia MF. Forty-six percent and 51% of patients in the VONJO and BAT treatment arms, respectively, had received prior ruxolitinib therapy. The median baseline hemoglobin level was 9.5 g/dL and 23% of patients were red blood cell (RBC) transfusion dependent at study entry. The median baseline platelet count was 55 × 10 9 /L; 45% of patients had a platelet count <50 × 10 9 /L. Patients had a baseline median spleen length of 14 cm assessed by magnetic resonance imaging (MRI) or computerized axial tomography (CAT).
Efficacy was established in patients who received VONJO 200 mg twice daily and had a platelet count <50 x 10 9 (N=31).
The most common agents used in the BAT treatment arm in patients with baseline platelet counts <50 × 10 9 /L (N=32) were ruxolitinib (39%), watchful waiting (32%), and hydroxyurea (26%).
Spleen Volume Reduction
The efficacy of VONJO in the treatment of patients with primary or secondary MF was established based upon the proportion of patients in the efficacy population receiving VONJO 200 mg twice daily or BAT achieving ≥35% spleen volume reduction from baseline to Week 24 as measured by magnetic resonance imaging or computed tomography. Efficacy results for spleen volume reduction in patients with a platelet count <50 × 10 9 /L are presented in Table 11
Table 11 Percentage of Patients Achieving ≥35% Reduction in Spleen Volume From Baseline to Week 24 in the Phase 3 Study, PERSIST-2 (Efficacy Population)
Patient Population | VONJO 200mg Twice Daily N=31 | Best Available Therapy N=32 |
Baseline Platelets <50 × 10 9 /L | 9 (29.0%) | 1 (3.1%) |
95% Confidence Interval (CI) | 14.2, 48.0 | 0.1, 16.2 |
Difference (VONJO-BAT) 95% CI | 25.9 (4.3,44.5) | |
A waterfall plot of the percentage of change in spleen volume from baseline to Week 24 is presented in Figure 1 for the PERSIST-2 patients with baseline platelet counts <50 × 10 9 /L. The median reduction in spleen volume for patients with a platelet count <50 × 10 9 /L was 27.3% for patients in the VONJO 200 mg twice daily group compared to 0.9% in the BAT group.
Figure 1 Waterfall Plot of Median Percent Change From Baseline in Spleen Volume at Week 24 in Patients With <50 × 109/L Platelet Counts in PERSIST-2 a
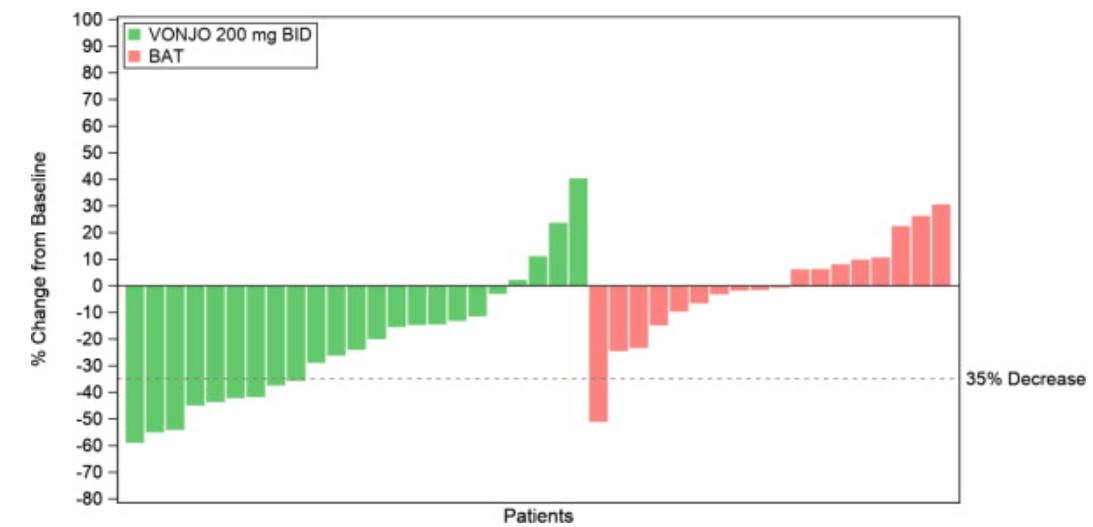
a Dropout rates in VONJO and BAT arms were 26% and 44%, respectively.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
VONJO is supplied in the following strength and package configuration:
| Strength | NDC Number | Description | Capsules per Bottle |
| 100 mg | 72482-100-12 | Hard, oblong, opaque, gelatin capsule with scarlet cap and gray body, printed with “Pacritinib 100 mg” on the cap and “C78837” on the body | 120 |
Storage
Store at room temperature, below 30°C (86°F). Keep the bottle tightly closed and protect from light. Store in original package. Dispense in original package or in a light-resistant container.
Mechanism of Action
Pacritinib is an oral kinase inhibitor with activity against wild type Janus associated kinase 2 (JAK2), mutant JAK2V617F, FMS-like tyrosine kinase 3 (FLT3), and interleukin 1 receptor associated kinase-1 (IRAK1) which contribute to signaling of a number of cytokines and growth factors that are important for hematopoiesis and immune function. Pacritinib is also an inhibitor of activin A receptor, type 1/activin receptor like-kinase 2 (ACVR1/ALK2).
MF is often associated with dysregulated JAK2 signaling. At clinically relevant concentrations, pacritinib does not inhibit JAK1. Pacritinib has higher inhibitory activity for JAK2 compared to JAK3 and tyrosine kinase 2 (TYK2). Pacritinib exhibits inhibitory activity against additional cellular kinases, such as colony stimulating factor 1 receptor (CSF1R), of which the clinical relevance in myelofibrosis is unknown.