Get your patient on Advair HFA (Fluticasone Propionate And Salmeterol Xinafoate)
Advair HFA patient education
Patient toolkit
Dosage & administration

Advair HFA prescribing information
INDICATIONS AND USAGE
ADVAIR HFA is indicated for treatment of asthma in adult and adolescent patients aged 12 years and older. ADVAIR HFA should be used for patients not adequately controlled on a long-term asthma control medication such as an inhaled corticosteroid (ICS) or whose disease warrants initiation of treatment with both an ICS and long-acting beta 2 -adrenergic agonist (LABA).
Limitations of Use
ADVAIR HFA is not indicated for the relief of acute bronchospasm.
DOSAGE AND ADMINISTRATION
Administration Information
ADVAIR HFA should be administered by the orally inhaled route only. After inhalation, rinse mouth with water without swallowing to help reduce the risk of oropharyngeal candidiasis.
Priming
Prime ADVAIR HFA before using for the first time by releasing 4 sprays into the air away from the face, shaking well for 5 seconds before each spray. In cases where the inhaler has not been used for more than 4 weeks or when it has been dropped, prime the inhaler again by releasing 2 sprays into the air away from the face, shaking well for 5 seconds before each spray. Avoid spraying in eyes.
Recommended Dosage
Adult and adolescent patients aged 12 years and older: 2 oral inhalations twice daily, approximately 12 hours apart.
The maximum recommended dosage is 2 inhalations of ADVAIR HFA 230 mcg/21 mcg twice daily.
General Dosing Recommendation
When choosing the starting dosage strength of ADVAIR HFA, consider the patients’ disease severity, based on their previous asthma therapy, including the ICS dosage, as well as the patients’ current control of asthma symptoms and risk of future exacerbation.
If asthma symptoms arise in the period between doses, an inhaled, short-acting beta 2 -agonist should be used for immediate relief.
Improvement in asthma control following inhaled administration of ADVAIR HFA can occur within 30 minutes of beginning treatment, although maximum benefit may not be achieved for 1 week or longer after starting treatment. Individual patients will experience a variable time to onset and degree of symptom relief.
For patients who do not respond adequately to the starting dosage after 2 weeks of therapy, replacing the current strength of ADVAIR HFA with a higher strength may provide additional improvement in asthma control.
If a previously effective dosage regimen fails to provide adequate improvement in asthma control, the therapeutic regimen should be reevaluated and additional therapeutic options (e.g., replacing the current strength of ADVAIR HFA with a higher strength, adding additional ICS, initiating oral corticosteroids) should be considered.
More frequent administration or a greater number of inhalations (more than 2 inhalations twice daily) of the prescribed strength of ADVAIR HFA is not recommended as some patients are more likely to experience adverse effects with higher doses of salmeterol. Patients using ADVAIR HFA should not use additional LABA for any reason. [See Warnings and Precautions (5.3 , 5.12 ).]
DOSAGE FORMS AND STRENGTHS
Inhalation aerosol: purple plastic inhaler with a light purple cap containing a pressurized metered-dose aerosol canister containing 60 or 120 metered inhalations and fitted with a counter.
- 45 mcg fluticasone propionate/21 mcg salmeterol from the mouthpiece per actuation
- 115 mcg fluticasone propionate/21 mcg salmeterol from the mouthpiece per actuation
- 230 mcg fluticasone propionate/21 mcg salmeterol from the mouthpiece per actuation
USE IN SPECIFIC POPULATIONS
Hepatic impairment: Monitor patients for signs of increased drug exposure. (8.6)
Pregnancy
Risk Summary
There are insufficient data on the use of ADVAIR HFA or individual monoproducts, fluticasone propionate and salmeterol xinafoate, in pregnant women. There are clinical considerations with the use of ADVAIR HFA in pregnant women. (See Clinical Considerations.) In animals, teratogenicity characteristic of corticosteroids, decreased fetal body weight and/or skeletal variations, in rats, mice, and rabbits, was observed with subcutaneously administered maternal toxic doses of fluticasone propionate less than the maximum recommended human daily inhaled dose (MRHDID) on a mcg/m 2 basis. (See Data.) However, fluticasone propionate administered via inhalation to rats decreased fetal body weight but did not induce teratogenicity at a maternal toxic dose less than the MRHDID on a mcg/m 2 basis. (See Data.) Experience with oral corticosteroids suggests that rodents are more prone to teratogenic effects from corticosteroids than humans. Oral administration of salmeterol to pregnant rabbits caused teratogenicity characteristic of beta-adrenoceptor stimulation at maternal doses approximately 25 times the MRHDID on an AUC basis. These adverse effects generally occurred at large multiples of the MRHDID when salmeterol was administered by the oral route to achieve high systemic exposures. No such effects occurred at an oral salmeterol dose approximately 10 times the MRHDID. (See Data.)
The estimated risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryofetal Risk: In women with poorly or moderately controlled asthma, there is an increased risk of several perinatal outcomes such as pre-eclampsia in the mother and prematurity, low birth weight, and small for gestational age in the neonate. Pregnant women should be closely monitored and medication adjusted as necessary to maintain optimal control of asthma.
Labor and Delivery: There are no human studies evaluating the effects of ADVAIR HFA during labor and delivery. Because of the potential for beta-agonist interference with uterine contractility, use of ADVAIR HFA during labor should be restricted to those patients in whom the benefits clearly outweigh the risks.
Data
Human Data: Fluticasone Propionate: Following inhaled administration, fluticasone propionate was detected in the neonatal cord blood after delivery.
Animal Data: Fluticasone Propionate and Salmeterol: In an embryofetal development study with pregnant rats that received the combination of subcutaneous administration of fluticasone propionate and oral administration of salmeterol at doses of 0/1,000; 30/0; 10/100; 30/1,000; and 100/10,000 mcg/kg/day (as fluticasone propionate/salmeterol) during the period of organogenesis, findings were generally consistent with the individual monoproducts and there was no exacerbation of expected fetal effects. Omphalocele, increased embryofetal deaths, decreased body weight, and skeletal variations were observed in rat fetuses in the presence of maternal toxicity when combining fluticasone propionate at a dose approximately equivalent to the MRHDID (on a mcg/m 2 basis at a maternal subcutaneous dose of 100 mcg/kg/day) and salmeterol at a dose approximately 1,200 times the MRHDID (on a mcg/m 2 basis at a maternal oral dose of 10,000 mcg/kg/day). The rat no observed adverse effect level (NOAEL) was observed when combining fluticasone propionate at a dose less than the MRHDID (on a mcg/m 2 basis at a maternal subcutaneous dose of 30 mcg/kg/day) and salmeterol at a dose approximately 120 times the MRHDID (on a mcg/m 2 basis at a maternal oral dose of 1,000 mcg/kg/day).
In an embryofetal development study with pregnant mice that received the combination of subcutaneous administration of fluticasone propionate and oral administration of salmeterol at doses of 0/1,400; 40/0; 10/200; 40/1,400; or 150/10,000 mcg/kg/day (as fluticasone propionate/salmeterol) during the period of organogenesis, findings were generally consistent with the individual monoproducts and there was no exacerbation of expected fetal effects. Cleft palate, fetal death, increased implantation loss, and delayed ossification were observed in mouse fetuses when combining fluticasone propionate at a dose approximately equivalent to the MRHDID (on a mcg/m 2 basis at a maternal subcutaneous dose of 150 mcg/kg/day) and salmeterol at a dose approximately 580 times the MRHDID (on a mcg/m 2 basis at a maternal oral dose of 10,000 mcg/kg/day). No developmental toxicity was observed at combination doses of fluticasone propionate up to approximately 0.2 times the MRHDID (on a mcg/m 2 basis at a maternal subcutaneous dose of 40 mcg/kg) and doses of salmeterol up to approximately 80 times the MRHDID (on a mcg/m 2 basis at a maternal oral dose of 1,400 mcg/kg).
Fluticasone Propionate: In embryofetal development studies with pregnant rats and mice dosed by the subcutaneous route throughout the period of organogenesis, fluticasone propionate was teratogenic in both species. Omphalocele, decreased body weight, and skeletal variations were observed in rat fetuses, in the presence of maternal toxicity, at a dose approximately equivalent to the MRHDID (on a mcg/m 2 basis with a maternal subcutaneous dose of 100 mcg/kg/day). The rat NOAEL was observed at approximately 0.3 times the MRHDID (on a mcg/m 2 basis with a maternal subcutaneous dose of 30 mcg/kg/day). Cleft palate and fetal skeletal variations were observed in mouse fetuses at a dose approximately 0.2 times the MRHDID (on a mcg/m 2 basis with a maternal subcutaneous dose of 45 mcg/kg/day). The mouse NOAEL was observed with a dose approximately 0.08 times the MRHDID (on a mcg/m 2 basis with a maternal subcutaneous dose of 15 mcg/kg/day).
In an embryofetal development study with pregnant rats dosed by the inhalation route throughout the period of organogenesis, fluticasone propionate produced decreased fetal body weights and skeletal variations, in the presence of maternal toxicity, at a dose approximately 0.3 times the MRHDID (on a mcg/m 2 basis with a maternal inhalation dose of 25.7 mcg/kg/day); however, there was no evidence of teratogenicity. The NOAEL was observed with a dose approximately 0.05 times the MRHDID (on a mcg/m 2 basis with a maternal inhalation dose of 5.5 mcg/kg/day).
In an embryofetal development study in pregnant rabbits that were dosed by the subcutaneous route throughout organogenesis, fluticasone propionate produced reductions of fetal body weights, in the presence of maternal toxicity, at doses less than the MRHDID (on a mcg/m 2 basis with a maternal subcutaneous dose of 0.57 mcg/kg/day). Teratogenicity was evident based upon a finding of cleft palate for 1 fetus at a dose approximately 0.09 times the MRHDID (on a mcg/m 2 basis with a maternal subcutaneous dose of 4 mcg/kg/day). The NOAEL was observed in rabbit fetuses with a dose approximately 0.002 times the MRHDID (on a mcg/m 2 basis with a maternal subcutaneous dose of 0.08 mcg/kg/day).
Fluticasone propionate crossed the placenta following subcutaneous administration to mice and rats and oral administration to rabbits.
In a pre- and post-natal development study in pregnant rats dosed by the subcutaneous route from late gestation through delivery and lactation (Gestation Day 17 to Postpartum Day 22), fluticasone propionate was not associated with decreases in pup body weight and had no effects on developmental landmarks, learning, memory, reflexes, or fertility at doses up to 0.5 times the MRHDID (on a mcg/m 2 basis with maternal subcutaneous doses up to 50 mcg/kg/day).
Salmeterol: In 3 embryofetal development studies, pregnant rabbits received oral administration of salmeterol at doses ranging from 100 to 10,000 mcg/kg/day during the period of organogenesis. In pregnant Dutch rabbits administered salmeterol doses approximately 25 times the MRHDID (on an AUC basis at maternal oral doses of 1,000 mcg/kg/day and higher), fetal toxic effects were observed characteristically resulting from beta-adrenoceptor stimulation. These included precocious eyelid openings, cleft palate, sternebral fusion, limb and paw flexures, and delayed ossification of the frontal cranial bones. No such effects occurred at a salmeterol dose approximately 10 times the MRHDID (on an AUC basis at a maternal oral dose of 600 mcg/kg/day). New Zealand White rabbits were less sensitive since only delayed ossification of the frontal cranial bones was seen at a salmeterol dose approximately 2,300 times the MRHDID (on a mcg/m 2 basis at a maternal oral dose of 10,000 mcg/kg/day).
In 2 embryofetal development studies, pregnant rats received salmeterol by oral administration at doses ranging from 100 to 10,000 mcg/kg/day during the period of organogenesis. Salmeterol produced no maternal toxicity or embryofetal effects at doses up to approximately 1,200 times the MRHDID (on a mcg/m 2 basis at maternal oral doses up to 10,000 mcg/kg/day).
In a peri- and post-natal development study in pregnant rats dosed by the oral route from late gestation through delivery and lactation, salmeterol at a dose approximately 1,200 times the MRHDID (on a mcg/m 2 basis with a maternal oral dose of 10,000 mcg/kg/day) was fetotoxic and decreased the fertility of survivors.
Salmeterol xinafoate crossed the placenta following oral administration to mice and rats.
Lactation
Risk Summary
There are no available data on the presence of fluticasone propionate or salmeterol in human milk, the effects on the breastfed child, or the effects on milk production. Other corticosteroids have been detected in human milk. However, fluticasone propionate and salmeterol concentrations in plasma after inhaled therapeutic doses are low and therefore concentrations in human breast milk are likely to be correspondingly low [see Clinical Pharmacology (12.3)] . The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ADVAIR HFA and any potential adverse effects on the breastfed child from ADVAIR HFA or from the underlying maternal condition.
Data
Animal Data: Subcutaneous administration of tritiated fluticasone propionate at a dose of 10 mcg/kg/day to lactating rats resulted in measurable levels in milk. Oral administration of salmeterol at a dose of 10,000 mcg/kg/day to lactating rats resulted in measurable levels in milk.
Pediatric Use
The safety and effectiveness of ADVAIR HFA have been established in pediatric patients aged 12 years and older. Use of ADVAIR HFA in this age group is supported by evidence from adequate and well-controlled studies in adults with additional data from thirty-eight (38) subjects aged 12 to 17 years were treated with ADVAIR HFA in U.S. pivotal clinical trials. Subjects in this age group demonstrated efficacy results similar to those observed in subjects aged 18 years and older. There were no obvious differences in the type or frequency of adverse events reported in this age group compared with subjects aged 18 years and older.
In a 12-week trial, the safety of ADVAIR HFA 45 mcg/21 mcg given as 2 inhalations twice daily was compared with that of fluticasone propionate 44 mcg HFA (FLOVENT HFA) 2 inhalations twice daily in 350 subjects aged 4 to 11 years with persistent asthma currently being treated with ICS. No new safety concerns were observed in children aged 4 to 11 years treated for 12 weeks with ADVAIR HFA 45 mcg/21 mcg compared with adults and adolescents aged 12 years and older. Common adverse reactions (≥3%) seen in children aged 4 to 11 years treated with ADVAIR HFA 45 mcg/21 mcg but not reported in the adult and adolescent clinical trials of ADVAIR HFA include: pyrexia, cough, pharyngolaryngeal pain, rhinitis, and sinusitis [see Adverse Reactions (6.1)] . This trial was not designed to assess the effect of salmeterol, a component of ADVAIR HFA, on asthma hospitalizations and death in subjects aged 4 to 11 years.
The pharmacokinetics and pharmacodynamic effect on serum cortisol of 21 days of treatment with ADVAIR HFA 45 mcg/21 mcg (2 inhalations twice daily with or without a spacer) or ADVAIR DISKUS 100 mcg/50 mcg (1 inhalation twice daily) was evaluated in a trial of 31 children aged 4 to 11 years with mild asthma. Systemic exposure to salmeterol xinafoate was similar for ADVAIR HFA, ADVAIR HFA delivered with a spacer, and ADVAIR DISKUS while the systemic exposure to fluticasone propionate was lower with ADVAIR HFA compared with that of ADVAIR HFA delivered with a spacer or ADVAIR DISKUS. There were reductions in serum cortisol from baseline in all treatment groups (14%, 22%, and 13% for ADVAIR HFA, ADVAIR HFA delivered with a spacer, and ADVAIR DISKUS, respectively) [see Clinical Pharmacology (12.2, 12.3)] .
The safety and effectiveness of ADVAIR HFA in pediatric patients younger than 12 years have not been established.
Effects on Growth
ICS, including fluticasone propionate, a component of ADVAIR HFA, may cause a reduction in growth velocity in children and adolescents [see Warnings and Precautions (5.14)] . The growth of pediatric patients receiving orally inhaled corticosteroids, including ADVAIR HFA, should be monitored.
A 52-week placebo-controlled trial to assess the potential growth effects of fluticasone propionate inhalation powder (FLOVENT ROTADISK) at 50 and 100 mcg twice daily was conducted in the U.S. in 325 prepubescent children (244 males and 81 females) aged 4 to 11 years. The mean growth velocities at 52 weeks observed in the intent-to-treat population were 6.32 cm/year in the placebo group (n = 76), 6.07 cm/year in the 50-mcg group (n = 98), and 5.66 cm/year in the 100-mcg group (n = 89). An imbalance in the proportion of children entering puberty between groups and a higher dropout rate in the placebo group due to poorly controlled asthma may be confounding factors in interpreting these data. A separate subset analysis of children who remained prepubertal during the trial revealed growth rates at 52 weeks of 6.10 cm/year in the placebo group (n = 57), 5.91 cm/year in the 50-mcg group (n = 74), and 5.67 cm/year in the 100-mcg group (n = 79). In children aged 8.5 years, the mean age of children in this trial, the range for expected growth velocity is: boys – 3 rd percentile = 3.8 cm/year, 50 th percentile = 5.4 cm/year, and 97 th percentile = 7.0 cm/year; girls – 3 rd percentile = 4.2 cm/year, 50 th percentile = 5.7 cm/year, and 97 th percentile = 7.3 cm/year. The clinical relevance of these growth data is not certain.
If a child or adolescent on any corticosteroid appears to have growth suppression, the possibility that he/she is particularly sensitive to this effect of corticosteroids should be considered. The potential growth effects of prolonged treatment should be weighed against the clinical benefits obtained. To minimize the systemic effects of orally inhaled corticosteroids, including ADVAIR HFA, each patient should be titrated to the lowest strength that effectively controls his/her asthma [see Dosage and Administration (2)] .
Geriatric Use
Clinical trials of ADVAIR HFA did not include sufficient numbers of subjects aged 65 years and older to determine whether older subjects respond differently than younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. In addition, as with other products containing beta 2 -agonists, special caution should be observed when using ADVAIR HFA in geriatric patients who have concomitant cardiovascular disease that could be adversely affected by beta 2 -agonists.
Hepatic Impairment
Formal pharmacokinetic studies using ADVAIR HFA have not been conducted in patients with hepatic impairment. However, since both fluticasone propionate and salmeterol are predominantly cleared by hepatic metabolism, impairment of liver function may lead to accumulation of fluticasone propionate and salmeterol in plasma. Therefore, patients with hepatic disease should be closely monitored.
Renal Impairment
Formal pharmacokinetic studies using ADVAIR HFA have not been conducted in patients with renal impairment.
CONTRAINDICATIONS
ADVAIR HFA is contraindicated in the following conditions:
WARNINGS AND PRECAUTIONS
- LABA monotherapy increases the risk of serious asthma-related events. (5.1)
- Do not initiate in acutely deteriorating asthma or to treat acute symptoms. (5.2)
- Do not use in combination with an additional medicine containing a LABA because of risk of overdose. (5.3)
- Candida albicans infection of the mouth and pharynx may occur. Monitor patients periodically. Advise the patient to rinse his/her mouth with water without swallowing after inhalation to help reduce the risk. (5.4 )
- Increased risk of pneumonia in patients with COPD. Monitor patients for signs and symptoms of pneumonia. (5.5 )
- Potential worsening of infections (e.g., existing tuberculosis; fungal, bacterial, viral, or parasitic infections; ocular herpes simplex). Use with caution in patients with these infections. More serious or even fatal course of chickenpox or measles can occur in susceptible patients. (5.6 )
- Risk of impaired adrenal function when transferring from systemic corticosteroids. Taper patients slowly from systemic corticosteroids if transferring to ADVAIR HFA. (5.7 )
- Hypercorticism and adrenal suppression may occur with very high dosages or at the regular dosage in susceptible individuals. If such changes occur, discontinue ADVAIR HFA slowly. (5.8 )
- If paradoxical bronchospasm occurs, discontinue ADVAIR HFA and institute alternative therapy. (5.10 )
- Use with caution in patients with cardiovascular or central nervous system disorders because of beta-adrenergic stimulation. (5.12 )
- Assess for decrease in bone mineral density initially and periodically thereafter. (5.13 )
- Monitor growth of pediatric patients. (5.14 )
- Glaucoma and cataracts may occur with long-term use of inhaled corticosteroids. Consider referral to an ophthalmologist in patients who develop ocular symptoms or use ADVAIR HFA long term. (5.15 )
- Be alert to eosinophilic conditions, hypokalemia, and hyperglycemia. (5.16 , 5.18 )
- Use with caution in patients with convulsive disorders, thyrotoxicosis, diabetes mellitus, and ketoacidosis. (5.17 )
Serious Asthma-Related Events – Hospitalizations, Intubations, Death
Use of LABA as monotherapy (without ICS) for asthma is associated with an increased risk of asthma-related death [see Salmeterol Multicenter Asthma Research Trial (SMART)] . Available data from controlled clinical trials also suggest that use of LABA as monotherapy increases the risk of asthma-related hospitalization in pediatric and adolescent patients. These findings are considered a class effect of LABA monotherapy. When LABA are used in fixed-dose combination with ICS, data from large clinical trials do not show a significant increase in the risk of serious asthma-related events (hospitalizations, intubations, death) compared with ICS alone (see Serious Asthma-Related Events with Inhaled Corticosteroid/Long-acting Beta 2 -adrenergic Agonists) .
Serious Asthma-Related Events with Inhaled Corticosteroid/Long-acting Beta 2 -adrenergic Agonists
Four (4) large, 26-week, randomized, double-blind, active-controlled clinical safety trials were conducted to evaluate the risk of serious asthma-related events when LABA were used in fixed‑dose combination with ICS compared with ICS alone in subjects with asthma. Three (3) trials included adult and adolescent subjects aged 12 years and older: 1 trial compared fluticasone propionate/salmeterol inhalation powder with fluticasone propionate inhalation powder, 1 trial compared mometasone furoate/formoterol with mometasone furoate, and 1 trial compared budesonide/formoterol with budesonide. The fourth trial included pediatric subjects aged 4 to 11 years and compared fluticasone propionate/salmeterol inhalation powder with fluticasone propionate inhalation powder. The primary safety endpoint for all 4 trials was serious asthma-related events (hospitalizations, intubations, death). A blinded adjudication committee determined whether events were asthma related.
The 3 adult and adolescent trials were designed to rule out a risk margin of 2.0, and the pediatric trial was designed to rule out a risk margin of 2.7. Each individual trial met its pre-specified objective and demonstrated non-inferiority of ICS/LABA to ICS alone. A meta-analysis of the 3 adult and adolescent trials did not show a significant increase in risk of a serious asthma-related event with ICS/LABA fixed-dose combination compared with ICS alone (Table 1). These trials were not designed to rule out all risk for serious asthma-related events with ICS/LABA compared with ICS.
| ICS = Inhaled Corticosteroid; LABA = Long-acting Beta 2 -adrenergic Agonist. a Randomized subjects who had taken at least 1 dose of study drug. Planned treatment used for analysis. b Estimated using a Cox proportional hazards model for time to first event with baseline hazards stratified by each of the 3 trials. c Number of subjects with event that occurred within 6 months after the first use of study drug or 7 days after the last date of study drug, whichever date was later. Subjects can have one or more events, but only the first event was counted for analysis. A single, blinded, independent adjudication committee determined whether events were asthma related. | |||
ICS/LABA (n = 17,537) a | ICS (n = 17,552) a | ICS/LABA vs. ICS Hazard Ratio (95% CI) b | |
Serious asthma-related event c | 116 | 105 | 1.10 (0.85, 1.44) |
Asthma-related death | 2 | 0 | |
Asthma-related intubation (endotracheal) | 1 | 2 | |
Asthma-related hospitalization (≥24-hour stay) | 115 | 105 | |
The pediatric safety trial included 6,208 pediatric subjects aged 4 to 11 years who received ICS/LABA (fluticasone propionate/salmeterol inhalation powder) or ICS (fluticasone propionate inhalation powder). In this trial, 27/3,107 (0.9%) subjects randomized to ICS/LABA and 21/3,101 (0.7%) subjects randomized to ICS experienced a serious asthma-related event. There were no asthma-related deaths or intubations. ICS/LABA did not show a significantly increased risk of a serious asthma-related event compared with ICS based on the pre-specified risk margin (2.7), with an estimated hazard ratio of time to first event of 1.29 (95% CI: 0.73, 2.27).
Salmeterol Multicenter Asthma Research Trial (SMART)
A 28-week, placebo-controlled, U.S. trial that compared the safety of salmeterol with placebo, each added to usual asthma therapy, showed an increase in asthma-related deaths in subjects receiving salmeterol (13/13,176 in subjects treated with salmeterol versus 3/13,179 in subjects treated with placebo; relative risk: 4.37 [95% CI: 1.25, 15.34]). Use of background ICS was not required in SMART. The increased risk of asthma‑related death is considered a class effect of LABA monotherapy.
Deterioration of Disease and Acute Episodes
ADVAIR HFA should not be initiated in patients during rapidly deteriorating or potentially life-threatening episodes of asthma. ADVAIR HFA has not been studied in subjects with acutely deteriorating asthma. The initiation of ADVAIR HFA in this setting is not appropriate.
Serious acute respiratory events, including fatalities, have been reported when salmeterol, a component of ADVAIR HFA, has been initiated in patients with significantly worsening or acutely deteriorating asthma. In most cases, these have occurred in patients with severe asthma (e.g., patients with a history of corticosteroid dependence, low pulmonary function, intubation, mechanical ventilation, frequent hospitalizations, previous life-threatening acute asthma exacerbations) and in some patients with acutely deteriorating asthma (e.g., patients with significantly increasing symptoms; increasing need for inhaled, short-acting beta 2 -agonists; decreasing response to usual medications; increasing need for systemic corticosteroids; recent emergency room visits; deteriorating lung function). However, these events have occurred in a few patients with less severe asthma as well. It was not possible from these reports to determine whether salmeterol contributed to these events.
Increasing use of inhaled, short-acting beta 2 -agonists is a marker of deteriorating asthma. In this situation, the patient requires immediate reevaluation with reassessment of the treatment regimen, giving special consideration to the possible need for replacing the current strength of ADVAIR HFA with a higher strength, adding additional ICS, or initiating systemic corticosteroids. Patients should not use more than 2 inhalations twice daily of ADVAIR HFA.
ADVAIR HFA should not be used for the relief of acute symptoms, i.e., as rescue therapy for the treatment of acute episodes of bronchospasm. ADVAIR HFA has not been studied in the relief of acute symptoms and extra doses should not be used for that purpose. Acute symptoms should be treated with an inhaled, short-acting beta 2 -agonist.
When beginning treatment with ADVAIR HFA, patients who have been taking oral or inhaled, short-acting beta 2 -agonists on a regular basis (e.g., 4 times a day) should be instructed to discontinue the regular use of these drugs.
Avoid Excessive Use of ADVAIR HFA and Avoid Use with Other Long-acting Beta 2 -agonists
ADVAIR HFA should not be used more often than recommended, at higher doses than recommended, or in conjunction with other medicines containing LABA, as an overdose may result. Clinically significant cardiovascular effects and fatalities have been reported in association with excessive use of inhaled sympathomimetic drugs. Patients using ADVAIR HFA should not use another medicine containing a LABA (e.g., salmeterol, formoterol fumarate, arformoterol tartrate, indacaterol) for any reason.
Oropharyngeal Candidiasis
In clinical trials, the development of localized infections of the mouth and pharynx with Candida albicans has occurred in subjects treated with ADVAIR HFA. When such an infection develops, it should be treated with appropriate local or systemic (i.e., oral) antifungal therapy while treatment with ADVAIR HFA continues, but at times therapy with ADVAIR HFA may need to be interrupted. Advise the patient to rinse his/her mouth with water without swallowing following inhalation to help reduce the risk of oropharyngeal candidiasis.
Pneumonia
Lower respiratory tract infections, including pneumonia, have been reported in patients with chronic obstructive pulmonary disease (COPD) following the inhaled administration of corticosteroids, including fluticasone propionate and ADVAIR DISKUS. In 2 replicate 1-year trials in 1,579 subjects with COPD, there was a higher incidence of pneumonia reported in subjects receiving ADVAIR DISKUS 250 mcg/50 mcg (7%) than in those receiving salmeterol 50 mcg (3%). The incidence of pneumonia in the subjects treated with ADVAIR DISKUS was higher in subjects older than 65 years (9%) compared with the incidence in subjects younger than 65 years (4%).
In a 3-year trial in 6,184 subjects with COPD, there was a higher incidence of pneumonia reported in subjects receiving ADVAIR DISKUS 500 mcg/50 mcg compared with placebo (16% with ADVAIR DISKUS 500 mcg/50 mcg, 14% with fluticasone propionate 500 mcg, 11% with salmeterol 50 mcg, and 9% with placebo). Similar to what was seen in the 1-year trials with ADVAIR DISKUS 250 mcg/50 mcg, the incidence of pneumonia was higher in subjects older than 65 years (18% with ADVAIR DISKUS 500 mcg/50 mcg versus 10% with placebo) compared with subjects younger than 65 years (14% with ADVAIR DISKUS 500 mcg/50 mcg versus 8% with placebo).
Immunosuppression and Risk of Infections
Persons who are using drugs that suppress the immune system are more susceptible to infections than healthy individuals. Chickenpox and measles, for example, can have a more serious or even fatal course in susceptible children or adults using corticosteroids. In such children or adults who have not had these diseases or been properly immunized, particular care should be taken to avoid exposure. How the dose, route, and duration of corticosteroid administration affect the risk of developing a disseminated infection is not known. The contribution of the underlying disease and/or prior corticosteroid treatment to the risk is also not known. If a patient is exposed to chickenpox, prophylaxis with varicella zoster immune globulin (VZIG) may be indicated. If a patient is exposed to measles, prophylaxis with pooled intramuscular immunoglobulin (IG) may be indicated. (See the respective package inserts for complete VZIG and IG prescribing information.) If chickenpox develops, treatment with antiviral agents may be considered.
ICS should be used with caution, if at all, in patients with active or quiescent tuberculosis infections of the respiratory tract; systemic fungal, bacterial, viral, or parasitic infections; or ocular herpes simplex.
Transferring Patients from Systemic Corticosteroid Therapy
HPA Suppression/Adrenal Insufficiency
Particular care is needed for patients who have been transferred from systemically active corticosteroids to ICS because deaths due to adrenal insufficiency have occurred in patients with asthma during and after transfer from systemic corticosteroids to less systemically available ICS. After withdrawal from systemic corticosteroids, a number of months are required for recovery of hypothalamic-pituitary-adrenal (HPA) function.
Patients who have been previously maintained on 20 mg or more of prednisone (or its equivalent) may be most susceptible, particularly when their systemic corticosteroids have been almost completely withdrawn. During this period of HPA suppression, patients may exhibit signs and symptoms of adrenal insufficiency when exposed to trauma, surgery, or infection (particularly gastroenteritis) or other conditions associated with severe electrolyte loss. Although ADVAIR HFA may control asthma symptoms during these episodes, in recommended doses it supplies less than normal physiological amounts of glucocorticoid systemically and does NOT provide the mineralocorticoid activity that is necessary for coping with these emergencies.
During periods of stress or a severe asthma attack, patients who have been withdrawn from systemic corticosteroids should be instructed to resume oral corticosteroids (in large doses) immediately and to contact their physicians for further instruction. These patients should also be instructed to carry a warning card indicating that they may need supplementary systemic corticosteroids during periods of stress or a severe asthma attack.
Patients requiring oral corticosteroids should be weaned slowly from systemic corticosteroid use after transferring to ADVAIR HFA. Prednisone reduction can be accomplished by reducing the daily prednisone dose by 2.5 mg on a weekly basis during therapy with ADVAIR HFA. Lung function (mean forced expiratory volume in 1 second [FEV 1 ] or morning peak expiratory flow [AM PEF]), beta-agonist use, and asthma symptoms should be carefully monitored during withdrawal of oral corticosteroids. In addition, patients should be observed for signs and symptoms of adrenal insufficiency, such as fatigue, lassitude, weakness, nausea and vomiting, and hypotension.
Unmasking of Allergic Conditions Previously Suppressed by Systemic Corticosteroids
Transfer of patients from systemic corticosteroid therapy to ADVAIR HFA may unmask allergic conditions previously suppressed by the systemic corticosteroid therapy (e.g., rhinitis, conjunctivitis, eczema, arthritis, eosinophilic conditions).
Corticosteroid Withdrawal Symptoms
During withdrawal from oral corticosteroids, some patients may experience symptoms of systemically active corticosteroid withdrawal (e.g., joint and/or muscular pain, lassitude, depression) despite maintenance or even improvement of respiratory function.
Hypercorticism and Adrenal Suppression
Fluticasone propionate, a component of ADVAIR HFA, will often help control asthma symptoms with less suppression of HPA function than therapeutically equivalent oral doses of prednisone. Since fluticasone propionate is absorbed into the circulation and can be systemically active at higher doses, the beneficial effects of ADVAIR HFA in minimizing HPA dysfunction may be expected only when recommended dosages are not exceeded and individual patients are titrated to the lowest effective dose. A relationship between plasma levels of fluticasone propionate and inhibitory effects on stimulated cortisol production has been shown after 4 weeks of treatment with fluticasone propionate inhalation aerosol. Since individual sensitivity to effects on cortisol production exists, physicians should consider this information when prescribing ADVAIR HFA.
Because of the possibility of significant systemic absorption of ICS in sensitive patients, patients treated with ADVAIR HFA should be observed carefully for any evidence of systemic corticosteroid effects. Particular care should be taken in observing patients postoperatively or during periods of stress for evidence of inadequate adrenal response.
It is possible that systemic corticosteroid effects such as hypercorticism and adrenal suppression (including adrenal crisis) may appear in a small number of patients who are sensitive to these effects. If such effects occur, ADVAIR HFA should be reduced slowly, consistent with accepted procedures for reducing systemic corticosteroids, and other treatments for management of asthma symptoms should be considered.
Drug Interactions with Strong Cytochrome P450 3A4 Inhibitors
The use of strong cytochrome P450 3A4 (CYP3A4) inhibitors (e.g., ritonavir, atazanavir, clarithromycin, indinavir, itraconazole, nefazodone, nelfinavir, saquinavir, ketoconazole, telithromycin) with ADVAIR HFA is not recommended because increased systemic corticosteroid and increased cardiovascular adverse effects may occur [see Drug Interactions (7.1 ), Clinical Pharmacology (12.3 )] .
Paradoxical Bronchospasm and Upper Airway Symptoms
As with other inhaled medicines, ADVAIR HFA can produce paradoxical bronchospasm, which may be life threatening. If paradoxical bronchospasm occurs following dosing with ADVAIR HFA, it should be treated immediately with an inhaled, short-acting bronchodilator; ADVAIR HFA should be discontinued immediately; and alternative therapy should be instituted. Upper airway symptoms of laryngeal spasm, irritation, or swelling, such as stridor and choking, have been reported in patients receiving ADVAIR HFA.
Hypersensitivity Reactions, including Anaphylaxis
Immediate hypersensitivity reactions (e.g., urticaria, angioedema, rash, bronchospasm, hypotension), including anaphylaxis, may occur after administration of ADVAIR HFA [see Contraindications (4 )] .
Cardiovascular and Central Nervous System Effects
Excessive beta-adrenergic stimulation has been associated with seizures, angina, hypertension or hypotension, tachycardia with rates up to 200 beats/min, arrhythmias, nervousness, headache, tremor, palpitation, nausea, dizziness, fatigue, malaise, and insomnia [see Overdosage (10)] . Therefore, ADVAIR HFA, like all products containing sympathomimetic amines, should be used with caution in patients with cardiovascular disorders, especially coronary insufficiency, cardiac arrhythmias, and hypertension.
Salmeterol, a component of ADVAIR HFA, can produce a clinically significant cardiovascular effect in some patients as measured by pulse rate, blood pressure, and/or symptoms. Although such effects are uncommon after administration of salmeterol at recommended doses, if they occur, the drug may need to be discontinued. In addition, beta-agonists have been reported to produce electrocardiogram (ECG) changes, such as flattening of the T wave, prolongation of the QTc interval, and ST segment depression. The clinical significance of these findings is unknown. Large doses of inhaled or oral salmeterol (12 to 20 times the recommended dose) have been associated with clinically significant prolongation of the QTc interval, which has the potential for producing ventricular arrhythmias. Fatalities have been reported in association with excessive use of inhaled sympathomimetic drugs.
Reduction in Bone Mineral Density
Decreases in bone mineral density (BMD) have been observed with long-term administration of products containing ICS. The clinical significance of small changes in BMD with regard to long-term consequences such as fracture is unknown. Patients with major risk factors for decreased bone mineral content, such as prolonged immobilization, family history of osteoporosis, postmenopausal status, tobacco use, advanced age, poor nutrition, or chronic use of drugs that can reduce bone mass (e.g., anticonvulsants, oral corticosteroids), should be monitored and treated with established standards of care.
2-Year Fluticasone Propionate Trial
A 2-year trial in 160 subjects (females aged 18 to 40 years, males 18 to 50) with asthma receiving chlorofluorocarbon (CFC)-propelled fluticasone propionate inhalation aerosol 88 or 440 mcg twice daily demonstrated no statistically significant changes in BMD at any time point (24, 52, 76, and 104 weeks of double-blind treatment) as assessed by dual-energy x-ray absorptiometry at lumbar regions L1 through L4.
Effect on Growth
Orally inhaled corticosteroids may cause a reduction in growth velocity when administered to pediatric patients. Monitor the growth of pediatric patients receiving ADVAIR HFA routinely (e.g., via stadiometry). To minimize the systemic effects of orally inhaled corticosteroids, including ADVAIR HFA, titrate each patient’s dosage to the lowest dosage that effectively controls his/her symptoms [see Dosage and Administration (2), Use in Specific Populations (8.4)] .
Glaucoma and Cataracts
Glaucoma, increased intraocular pressure, and cataracts have been reported in patients with asthma following the long-term administration of ICS, including fluticasone propionate, a component of ADVAIR HFA. Consider referral to an ophthalmologist in patients who develop ocular symptoms or use ADVAIR HFA long term [see Adverse Reactions (6)] .
Eosinophilic Conditions and Churg-Strauss Syndrome
In rare cases, patients on inhaled fluticasone propionate, a component of ADVAIR HFA, may present with systemic eosinophilic conditions. Some of these patients have clinical features of vasculitis consistent with Churg-Strauss syndrome, a condition that is often treated with systemic corticosteroid therapy. These events usually, but not always, have been associated with the reduction and/or withdrawal of oral corticosteroid therapy following the introduction of fluticasone propionate. Cases of serious eosinophilic conditions have also been reported with other ICS in this clinical setting. Physicians should be alert to eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy presenting in their patients. A causal relationship between fluticasone propionate and these underlying conditions has not been established.
Coexisting Conditions
ADVAIR HFA, like all medicines containing sympathomimetic amines, should be used with caution in patients with convulsive disorders or thyrotoxicosis and in those who are unusually responsive to sympathomimetic amines. Large doses of the related beta 2 -adrenoceptor agonist albuterol, when administered intravenously, have been reported to aggravate preexisting diabetes mellitus and ketoacidosis.
Hypokalemia and Hyperglycemia
Beta-adrenergic agonist medicines may produce significant hypokalemia in some patients, possibly through intracellular shunting, which has the potential to produce adverse cardiovascular effects [see Clinical Pharmacology (12.2)] . The decrease in serum potassium is usually transient, not requiring supplementation. Clinically significant changes in blood glucose and/or serum potassium were seen infrequently during clinical trials with ADVAIR HFA at recommended doses.
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious asthma-related events – hospitalizations, intubations, death [see Warnings and Precautions (5.1)]
- Oropharyngeal candidiasis [see Warnings and Precautions (5.4)]
- Pneumonia in patients with COPD [see Warnings and Precautions (5.5)]
- Immunosuppression and risk of infections [see Warnings and Precautions (5.6)]
- Hypercorticism and adrenal suppression [see Warnings and Precautions (5.8)]
- Cardiovascular and central nervous system effects [see Warnings and Precautions (5.12)]
- Reduction in bone mineral density [see Warnings and Precautions (5.13)]
- Growth effects [see Warnings and Precautions (5.14)]
- Glaucoma and cataracts [see Warnings and Precautions (5.15)]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult and Adolescent Subjects Aged 12 Years and Older
The incidence of adverse reactions associated with ADVAIR HFA in Table 2 is based upon two 12-week, placebo-controlled, U.S. clinical trials (Trials 1 and 3) and 1 active-controlled 12-week U.S. clinical trial (Trial 2). A total of 1,008 adult and adolescent subjects with asthma (556 females and 452 males) previously treated with albuterol alone, salmeterol, or ICS were treated twice daily with 2 inhalations of ADVAIR HFA 45 mcg/21 mcg or ADVAIR HFA 115 mcg/21 mcg, fluticasone propionate CFC inhalation aerosol (44- or 110-mcg doses), salmeterol CFC inhalation aerosol 21 mcg, or placebo HFA inhalation aerosol. The average duration of exposure was 71 to 81 days in the active treatment groups compared with 51 days in the placebo group.
| ADVAIR HFA | Fluticasone Propionate CFC Inhalation Aerosol | Salmeterol CFC Inhalation Aerosol | Placebo HFA Inhalation Aerosol | ||
|
|
|
|
|
| |
Ear, nose, and throat | ||||||
Upper respiratory tract infection | 16 | 24 | 13 | 15 | 17 | 13 |
Throat irritation | 9 | 7 | 12 | 13 | 9 | 7 |
Upper respiratory inflammation | 4 | 4 | 3 | 7 | 5 | 3 |
Hoarseness/dysphonia | 3 | 1 | 2 | 0 | 1 | 0 |
Lower respiratory | ||||||
Viral respiratory infection | 3 | 5 | 4 | 5 | 3 | 4 |
Neurology | ||||||
Headache | 21 | 15 | 24 | 16 | 20 | 11 |
Dizziness | 4 | 1 | 1 | 0 | <1 | 0 |
Gastrointestinal | ||||||
Nausea and vomiting | 5 | 3 | 4 | 2 | 2 | 3 |
Viral gastrointestinal infection | 4 | 2 | 2 | 0 | 1 | 2 |
Gastrointestinal signs and symptoms | 3 | 2 | 2 | 1 | 1 | 1 |
Musculoskeletal | ||||||
Musculoskeletal pain | 5 | 7 | 8 | 2 | 4 | 4 |
Muscle pain | 4 | 1 | 1 | 1 | 3 | <1 |
The incidence of common adverse reactions reported in Trial 4, a 12-week non-U.S. clinical trial in 509 subjects previously treated with ICS who were treated twice daily with 2 inhalations of ADVAIR HFA 230 mcg/21 mcg, fluticasone propionate CFC inhalation aerosol 220 mcg, or 1 inhalation of ADVAIR DISKUS 500 mcg/50 mcg was similar to the incidences reported in Table 2.
Additional Adverse Reactions
Other adverse reactions not previously listed, whether considered drug-related or not by the investigators, that occurred in the groups receiving ADVAIR HFA with an incidence of 1% to 3% and that occurred at a greater incidence than with placebo include the following: tachycardia, arrhythmias, myocardial infarction, postoperative complications, wounds and lacerations, soft tissue injuries, ear signs and symptoms, rhinorrhea/postnasal drip, epistaxis, nasal congestion/blockage, laryngitis, unspecified oropharyngeal plaques, dryness of nose, weight gain, allergic eye disorders, eye edema and swelling, gastrointestinal discomfort and pain, dental discomfort and pain, candidiasis mouth/throat, hyposalivation, gastrointestinal infections, disorders of hard tissue of teeth, abdominal discomfort and pain, oral abnormalities, arthralgia and articular rheumatism, muscle cramps and spasms, musculoskeletal inflammation, bone and skeletal pain, muscle injuries, sleep disorders, migraines, allergies and allergic reactions, viral infections, bacterial infections, candidiasis unspecified site, congestion, inflammation, bacterial reproductive infections, lower respiratory signs and symptoms, lower respiratory infections, lower respiratory hemorrhage, eczema, dermatitis and dermatosis, urinary infections.
Laboratory Test Abnormalities
In Trial 3, there were more reports of hyperglycemia among adults and adolescents receiving ADVAIR HFA, but this was not seen in Trials 1 and 2.
Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been identified during postapproval use of any formulation of ADVAIR, fluticasone propionate, and/or salmeterol regardless of indication. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These events have been chosen for inclusion due to either their seriousness, frequency of reporting, or causal connection to ADVAIR, fluticasone propionate, and/or salmeterol or a combination of these factors.
Cardiovascular
Arrhythmias (including atrial fibrillation, extrasystoles, supraventricular tachycardia), hypertension, ventricular tachycardia.
Ear, Nose, and Throat
Aphonia, earache, facial and oropharyngeal edema, paranasal sinus pain, rhinitis, throat soreness, tonsillitis.
Endocrine and Metabolic
Cushing’s syndrome, Cushingoid features, growth velocity reduction in children/adolescents, hypercorticism, osteoporosis.
Eye
Cataracts, glaucoma.
Gastrointestinal
Dyspepsia, xerostomia.
Hepatobiliary Tract and Pancreas
Abnormal liver function tests.
Immune System
Immediate and delayed hypersensitivity reactions, including rash and rare events of angioedema, bronchospasm, and anaphylaxis.
Infections and Infestations
Esophageal candidiasis.
Musculoskeletal
Back pain, myositis.
Neurology
Paresthesia, restlessness.
Non-Site Specific
Fever, pallor.
Psychiatry
Agitation, aggression, anxiety, depression. Behavioral changes, including hyperactivity and irritability, have been reported very rarely and primarily in children.
Respiratory
Asthma; asthma exacerbation; chest congestion; chest tightness; cough; dyspnea; immediate bronchospasm; influenza; paradoxical bronchospasm; tracheitis; wheezing; pneumonia; reports of upper respiratory symptoms of laryngeal spasm, irritation, or swelling such as stridor or choking.
Skin
Contact dermatitis, contusions, ecchymoses, photodermatitis, pruritus.
Urogenital
Dysmenorrhea, irregular menstrual cycle, pelvic inflammatory disease, vaginal candidiasis, vaginitis, vulvovaginitis.
DRUG INTERACTIONS
ADVAIR HFA has been used concomitantly with other drugs, including short-acting beta 2 -agonists, methylxanthines, and nasal corticosteroids, commonly used in patients with asthma without adverse drug reactions [see Clinical Pharmacology (12.2)] . No formal drug interaction trials have been performed with ADVAIR HFA.
Inhibitors of Cytochrome P450 3A4
Fluticasone propionate and salmeterol, the individual components of ADVAIR HFA, are substrates of CYP3A4. The use of strong CYP3A4 inhibitors (e.g., ritonavir, atazanavir, clarithromycin, indinavir, itraconazole, nefazodone, nelfinavir, saquinavir, ketoconazole, telithromycin) with ADVAIR HFA is not recommended because increased systemic corticosteroid and increased cardiovascular adverse effects may occur.
Ritonavir
Fluticasone Propionate: A drug interaction trial with fluticasone propionate aqueous nasal spray in healthy subjects has shown that ritonavir (a strong CYP3A4 inhibitor) can significantly increase plasma fluticasone propionate exposure, resulting in significantly reduced serum cortisol concentrations [see Clinical Pharmacology (12.3)] . During postmarketing use, there have been reports of clinically significant drug interactions in patients receiving fluticasone propionate and ritonavir, resulting in systemic corticosteroid effects including Cushing’s syndrome and adrenal suppression.
Ketoconazole
Fluticasone Propionate: Coadministration of orally inhaled fluticasone propionate (1,000 mcg) and ketoconazole (200 mg once daily) resulted in a 1.9-fold increase in plasma fluticasone propionate exposure and a 45% decrease in plasma cortisol area under the curve (AUC), but had no effect on urinary excretion of cortisol.
Salmeterol: In a drug interaction trial in 20 healthy subjects, coadministration of inhaled salmeterol (50 mcg twice daily) and oral ketoconazole (400 mg once daily) for 7 days resulted in greater systemic exposure to salmeterol (AUC increased 16-fold and C max increased 1.4-fold). Three (3) subjects were withdrawn due to beta 2 -agonist side effects (2 with prolonged QTc and 1 with palpitations and sinus tachycardia). Although there was no statistical effect on the mean QTc, coadministration of salmeterol and ketoconazole was associated with more frequent increases in QTc duration compared with salmeterol and placebo administration.
Monoamine Oxidase Inhibitors and Tricyclic Antidepressants
ADVAIR HFA should be administered with extreme caution to patients being treated with monoamine oxidase inhibitors or tricyclic antidepressants, or within 2 weeks of discontinuation of such agents, because the action of salmeterol, a component of ADVAIR HFA, on the vascular system may be potentiated by these agents.
Beta-adrenergic Receptor Blocking Agents
Beta-blockers not only block the pulmonary effect of beta-agonists, such as salmeterol, a component of ADVAIR HFA, but may also produce severe bronchospasm in patients with asthma. Therefore, patients with asthma should not normally be treated with beta-blockers. However, under certain circumstances, there may be no acceptable alternatives to the use of beta-adrenergic blocking agents for these patients; cardioselective beta-blockers could be considered, although they should be administered with caution.
Non–Potassium-Sparing Diuretics
The ECG changes and/or hypokalemia that may result from the administration of non–potassium-sparing diuretics (such as loop or thiazide diuretics) can be acutely worsened by beta-agonists, such as salmeterol, a component of ADVAIR HFA, especially when the recommended dose of the beta-agonist is exceeded. Although the clinical significance of these effects is not known, caution is advised in the coadministration of ADVAIR HFA with non–potassium-sparing diuretics.
DESCRIPTION
ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, and ADVAIR HFA 230 mcg/21 mcg are combinations of fluticasone propionate and salmeterol xinafoate.
One active component of ADVAIR HFA is fluticasone propionate, a corticosteroid having the chemical name S- (fluoromethyl) 6α,9-difluoro-11β,17-dihydroxy-16α-methyl-3-oxoandrosta-1,4-diene-17β-carbothioate, 17-propionate and the following chemical structure:
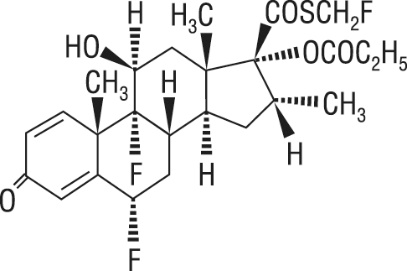
Fluticasone propionate is a white powder with a molecular weight of 500.6, and the empirical formula is C 25 H 31 F 3 O 5 S. It is practically insoluble in water, freely soluble in dimethyl sulfoxide and dimethylformamide, and slightly soluble in methanol and 95% ethanol.
The other active component of ADVAIR HFA is salmeterol xinafoate, a beta 2 -adrenergic bronchodilator. Salmeterol xinafoate is the racemic form of the 1-hydroxy-2-naphthoic acid salt of salmeterol. It has the chemical name 4-hydroxy-α 1 -[[[6-(4-phenylbutoxy)hexyl]amino]methyl]-1,3-benzenedimethanol, 1-hydroxy-2-naphthalenecarboxylate and the following chemical structure:
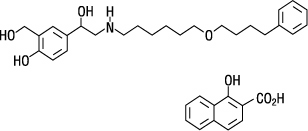
Salmeterol xinafoate is a white powder with a molecular weight of 603.8, and the empirical formula is C 25 H 37 NO 4 •C 11 H 8 O 3 . It is freely soluble in methanol; slightly soluble in ethanol, chloroform, and isopropanol; and sparingly soluble in water.
ADVAIR HFA is a purple plastic inhaler with a light purple cap containing a pressurized metered-dose aerosol canister fitted with a counter. Each canister contains a microcrystalline suspension of micronized fluticasone propionate and micronized salmeterol xinafoate in propellant HFA-134a (1,1,1,2-tetrafluoroethane). It contains no other excipients.
After priming, each actuation of the inhaler delivers 50, 125, or 250 mcg of fluticasone propionate and 25 mcg of salmeterol in 75 mg of suspension from the valve. Each actuation delivers 45, 115, or 230 mcg of fluticasone propionate and 21 mcg of salmeterol from the actuator. Twenty-one micrograms (21 mcg) of salmeterol base is equivalent to 30.45 mcg of salmeterol xinafoate. The actual amount of drug delivered to the lung will depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system.
Prime ADVAIR HFA before using for the first time by releasing 4 sprays into the air away from the face, shaking well for 5 seconds before each spray. In cases where the inhaler has not been used for more than 4 weeks or when it has been dropped, prime the inhaler again by releasing 2 sprays into the air away from the face, shaking well for 5 seconds before each spray. Avoid spraying in eyes.
CLINICAL PHARMACOLOGY
Mechanism of Action
ADVAIR HFA
ADVAIR HFA contains both fluticasone propionate and salmeterol. The mechanisms of action described below for the individual components apply to ADVAIR HFA. These drugs represent 2 different classes of medications (a synthetic corticosteroid and a LABA) that have different effects on clinical, physiologic, and inflammatory indices of asthma.
Fluticasone Propionate
Fluticasone propionate is a synthetic trifluorinated corticosteroid with anti-inflammatory activity. Fluticasone propionate has been shown in vitro to exhibit a binding affinity for the human glucocorticoid receptor that is 18 times that of dexamethasone, almost twice that of beclomethasone-17-monopropionate (BMP), the active metabolite of beclomethasone dipropionate, and over 3 times that of budesonide. Data from the McKenzie vasoconstrictor assay in man are consistent with these results. The clinical significance of these findings is unknown.
Inflammation is an important component in the pathogenesis of asthma. Corticosteroids have been shown to have a wide range of actions on multiple cell types (e.g., mast cells, eosinophils, neutrophils, macrophages, lymphocytes) and mediators (e.g., histamine, eicosanoids, leukotrienes, cytokines) involved in inflammation. These anti-inflammatory actions of corticosteroids contribute to their efficacy in asthma.
Salmeterol Xinafoate
Salmeterol is a selective LABA. In vitro studies show salmeterol to be at least 50 times more selective for beta 2 ‑adrenoceptors than albuterol. Although beta 2 -adrenoceptors are the predominant adrenergic receptors in bronchial smooth muscle and beta 1 -adrenoceptors are the predominant receptors in the heart, there are also beta 2 -adrenoceptors in the human heart comprising 10% to 50% of the total beta-adrenoceptors. The precise function of these receptors has not been established, but their presence raises the possibility that even selective beta 2 -agonists may have cardiac effects.
The pharmacologic effects of beta 2 -adrenoceptor agonist drugs, including salmeterol, are at least in part attributable to stimulation of intracellular adenyl cyclase, the enzyme that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3′,5′-adenosine monophosphate (cyclic AMP). Increased cyclic AMP levels cause relaxation of bronchial smooth muscle and inhibition of release of mediators of immediate hypersensitivity from cells, especially from mast cells.
In vitro tests show that salmeterol is a potent and long-lasting inhibitor of the release of mast cell mediators, such as histamine, leukotrienes, and prostaglandin D 2 , from human lung. Salmeterol inhibits histamine-induced plasma protein extravasation and inhibits platelet-activating factor–induced eosinophil accumulation in the lungs of guinea pigs when administered by the inhaled route. In humans, single doses of salmeterol administered via inhalation aerosol attenuate allergen-induced bronchial hyper-responsiveness.
Pharmacodynamics
ADVAIR HFA
Healthy Subjects: Cardiovascular Effects: Since systemic pharmacodynamic effects of salmeterol are not normally seen at the therapeutic dose, higher doses were used to produce measurable effects. Four (4) placebo-controlled crossover trials were conducted with healthy subjects: (1) a cumulative-dose trial using 42 to 336 mcg of salmeterol CFC inhalation aerosol given alone or as ADVAIR HFA 115 mcg/21 mcg, (2) a single-dose trial using 4 inhalations of ADVAIR HFA 230 mcg/21 mcg, salmeterol CFC inhalation aerosol 21 mcg, or fluticasone propionate CFC inhalation aerosol 220 mcg, (3) a single-dose trial using 8 inhalations of ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, or ADVAIR HFA 230 mcg/21 mcg, and (4) a single-dose trial using 4 inhalations of ADVAIR HFA 230 mcg/21 mcg; 2 inhalations of ADVAIR DISKUS 500 mcg/50 mcg; 4 inhalations of fluticasone propionate CFC inhalation aerosol 220 mcg; or 1,010 mcg of fluticasone propionate given intravenously. In these trials pulse rate, blood pressure, QTc interval, glucose, and/or potassium were measured. Comparable or lower effects were observed for ADVAIR HFA compared with ADVAIR DISKUS or salmeterol alone. The effect of salmeterol on pulse rate and potassium was not altered by the presence of different amounts of fluticasone propionate in ADVAIR HFA.
Hypothalamic-Pituitary-Adrenal Axis Effects: The potential effect of salmeterol on the effects of fluticasone propionate on the HPA axis was also evaluated in 3 of these trials. Compared with fluticasone propionate CFC inhalation aerosol, ADVAIR HFA had less effect on 24-hour urinary cortisol excretion and less or comparable effect on 24-hour serum cortisol. In these crossover trials in healthy subjects, ADVAIR HFA and ADVAIR DISKUS had similar effects on urinary and serum cortisol.
Subjects with Asthma: Cardiovascular Effects: In clinical trials with ADVAIR HFA in adult and adolescent subjects aged 12 years and older with asthma, systemic pharmacodynamic effects of salmeterol (pulse rate, blood pressure, QTc interval, potassium, and glucose) were similar to or slightly lower in patients treated with ADVAIR HFA compared with patients treated with salmeterol CFC inhalation aerosol 21 mcg. In 61 adult and adolescent subjects with asthma given ADVAIR HFA (45 mcg/21 mcg or 115 mcg/21 mcg), continuous 24-hour electrocardiographic monitoring was performed after the first dose and after 12 weeks of twice-daily therapy, and no clinically significant dysrhythmias were noted.
The effect of 21 days of treatment with ADVAIR HFA 45 mcg/21 mcg (2 inhalations twice daily with or without a spacer) or ADVAIR DISKUS 100 mcg/50 mcg (1 inhalation twice daily) was evaluated in 31 children aged 4 to 11 years with mild asthma. There were no notable changes from baseline for QTc, heart rate, or systolic and diastolic blood pressure.
Hypothalamic-Pituitary-Adrenal Axis Effects: A 4-way crossover trial in 13 subjects with asthma compared pharmacodynamics at steady state following 4 weeks of twice-daily treatment with 2 inhalations of ADVAIR HFA 115 mcg/21 mcg, 1 inhalation of ADVAIR DISKUS 250 mcg/50 mcg, 2 inhalations of fluticasone propionate HFA inhalation aerosol 110 mcg, and placebo. No significant differences in serum cortisol AUC were observed between active treatments and placebo. Mean 12-hour serum cortisol AUC ratios comparing active treatment with placebo ranged from 0.9 to 1.2. No statistically or clinically significant increases in heart rate or QTc interval were observed for any active treatment compared with placebo.
In a 12-week trial in adult and adolescent subjects with asthma, ADVAIR HFA 115 mcg/21 mcg was compared with the individual components, fluticasone propionate CFC inhalation aerosol 110 mcg and salmeterol CFC inhalation aerosol 21 mcg, and placebo [see Clinical Studies (14.1)] . All treatments were administered as 2 inhalations twice daily. After 12 weeks of treatment with these therapeutic doses, the geometric mean ratio of urinary cortisol excretion compared with baseline was 0.9 for ADVAIR HFA and fluticasone propionate and 1.0 for placebo and salmeterol. In addition, the ability to increase cortisol production in response to stress, as assessed by 30-minute cosyntropin stimulation in 23 to 32 subjects per treatment group, remained intact for the majority of subjects and was similar across treatments. Three (3) subjects who received ADVAIR HFA 115 mcg/21 mcg had an abnormal response (peak serum cortisol <18 mcg/dL) after dosing, compared with 1 subject who received placebo, 2 subjects who received fluticasone propionate 110 mcg, and 1 subject who received salmeterol.
In another 12-week trial in adult and adolescent subjects with asthma, ADVAIR HFA 230 mcg/21 mcg (2 inhalations twice daily) was compared with ADVAIR DISKUS 500 mcg/50 mcg (1 inhalation twice daily) and fluticasone propionate CFC inhalation aerosol 220 mcg (2 inhalations twice daily) [see Clinical Studies (14.1)] . The geometric mean ratio of 24‑hour urinary cortisol excretion at week 12 compared with baseline was 0.9 for all 3 treatment groups.
The effect of 21 days of treatment with ADVAIR HFA 45 mcg/21 mcg (2 inhalations twice daily with or without a spacer) or ADVAIR DISKUS 100 mcg/50 mcg (1 inhalation twice daily) on serum cortisol was evaluated in 31 children aged 4 to 11 years with mild asthma. There were reductions in serum cortisol from baseline in all treatment groups (14%, 22%, and 13% for ADVAIR HFA, ADVAIR HFA with spacer, and ADVAIR DISKUS, respectively).
Other Fluticasone Propionate Products
Subjects with Asthma: Hypothalamic-Pituitary-Adrenal Axis Effects: In clinical trials with fluticasone propionate inhalation powder using dosages up to and including 250 mcg twice daily, occasional abnormal short cosyntropin tests (peak serum cortisol <18 mcg/dL assessed by radioimmunoassay) were noted both in subjects receiving fluticasone propionate and in subjects receiving placebo. The incidence of abnormal tests at 500 mcg twice daily was greater than placebo. In a 2-year trial carried out with the DISKHALER inhalation device in 64 subjects with mild, persistent asthma (mean FEV 1 91% of predicted) randomized to fluticasone propionate 500 mcg twice daily or placebo, no subject receiving fluticasone propionate had an abnormal response to 6-hour cosyntropin infusion (peak serum cortisol <18 mcg/dL). With a peak cortisol threshold of <35 mcg/dL, 1 subject receiving fluticasone propionate (4%) had an abnormal response at 1 year; repeat testing at 18 months and 2 years was normal. Another subject receiving fluticasone propionate (5%) had an abnormal response at 2 years. No subject on placebo had an abnormal response at 1 or 2 years.
Other Salmeterol Xinafoate Products
Subjects with Asthma: Cardiovascular Effects: Inhaled salmeterol, like other beta-adrenergic agonist drugs, can produce dose-related cardiovascular effects and effects on blood glucose and/or serum potassium [see Warnings and Precautions (5.12, 5.18)] . The cardiovascular effects (heart rate, blood pressure) associated with salmeterol inhalation aerosol occur with similar frequency, and are of similar type and severity, as those noted following albuterol administration.
The effects of rising inhaled doses of salmeterol and standard inhaled doses of albuterol were studied in volunteers and in subjects with asthma. Salmeterol doses up to 84 mcg administered as inhalation aerosol resulted in heart rate increases of 3 to 16 beats/min, about the same as albuterol dosed at 180 mcg by inhalation aerosol (4 to 10 beats/min). In 2 double‑blind asthma trials, subjects receiving either 42 mcg of salmeterol inhalation aerosol twice daily (n = 81) or 180 mcg of albuterol inhalation aerosol 4 times daily (n = 80) underwent continuous electrocardiographic monitoring during four 24-hour periods; no clinically significant dysrhythmias were noted.
Concomitant Use of ADVAIR HFA with Other Respiratory Medicines
Short-acting Beta 2 -agonists: In three 12-week U.S. clinical trials, the mean daily need for additional beta 2 -agonist use in 277 subjects receiving ADVAIR HFA was approximately 1.2 inhalations/day and ranged from 0 to 9 inhalations/day. Two percent (2%) of subjects receiving ADVAIR HFA in these trials averaged 6 or more inhalations per day over the course of the 12-week trials. No increase in frequency of cardiovascular adverse events was observed among subjects who averaged 6 or more inhalations per day.
Methylxanthines: The concurrent use of intravenously or orally administered methylxanthines (e.g., aminophylline, theophylline) by subjects receiving ADVAIR HFA has not been completely evaluated. In five 12-week clinical trials (3 U.S. and 2 non-U.S.), 45 subjects receiving ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, or ADVAIR HFA 230 mcg/21 mcg twice daily concurrently with a theophylline product had adverse event rates similar to those in 577 subjects receiving ADVAIR HFA without theophylline.
Fluticasone Propionate Nasal Spray: In subjects receiving ADVAIR HFA in three 12-week U.S. clinical trials, no difference in the profile of adverse events or HPA axis effects was noted between subjects receiving FLONASE (fluticasone propionate) Nasal Spray, 50 mcg concurrently (n = 89) and those who were not (n = 192).
Pharmacokinetics
Absorption
Fluticasone Propionate: Healthy Subjects: Fluticasone propionate acts locally in the lung; therefore, plasma levels do not predict therapeutic effect. Trials using oral dosing of labeled and unlabeled drug have demonstrated that the oral systemic bioavailability of fluticasone propionate is negligible (<1%), primarily due to incomplete absorption and presystemic metabolism in the gut and liver. In contrast, the majority of the fluticasone propionate delivered to the lung is systemically absorbed.
Three (3) single-dose, placebo-controlled, crossover trials were conducted in healthy subjects: (1) a trial using 4 inhalations of ADVAIR HFA 230 mcg/21 mcg, salmeterol CFC inhalation aerosol 21 mcg, or fluticasone propionate CFC inhalation aerosol 220 mcg, (2) a trial using 8 inhalations of ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, or ADVAIR HFA 230 mcg/21 mcg, and (3) a trial using 4 inhalations of ADVAIR HFA 230 mcg/21 mcg; 2 inhalations of ADVAIR DISKUS 500 mcg/50 mcg; 4 inhalations of fluticasone propionate CFC inhalation aerosol 220 mcg; or 1,010 mcg of fluticasone propionate given intravenously. Peak plasma concentrations of fluticasone propionate were achieved in 0.33 to 1.5 hours and those of salmeterol were achieved in 5 to 10 minutes.
Peak plasma concentrations of fluticasone propionate (N = 20 subjects) following 8 inhalations of ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, and ADVAIR HFA 230 mcg/21 mcg averaged 41, 108, and 173 pg/mL, respectively.
Systemic exposure (N = 20 subjects) from 4 inhalations of ADVAIR HFA 230 mcg/21 mcg was 53% of the value from the individual inhaler for fluticasone propionate CFC inhalation aerosol and 42% of the value from the individual inhaler for salmeterol CFC inhalation aerosol. Peak plasma concentrations from ADVAIR HFA for fluticasone propionate (86 versus 120 pg/mL) and salmeterol (170 versus 510 pg/mL) were significantly lower compared with individual inhalers.
In 15 healthy subjects, systemic exposure to fluticasone propionate from 4 inhalations of ADVAIR HFA 230 mcg/21 mcg (920/84 mcg) and 2 inhalations of ADVAIR DISKUS 500 mcg/50 mcg (1,000/100 mcg) was similar between the 2 inhalers (i.e., 799 versus 832 pg•h/mL, respectively), but approximately half the systemic exposure from 4 inhalations of fluticasone propionate CFC inhalation aerosol 220 mcg (880 mcg, AUC = 1,543 pg•h/mL). Similar results were observed for peak fluticasone propionate plasma concentrations (186 and 182 pg/mL from ADVAIR HFA and ADVAIR DISKUS, respectively, and 307 pg/mL from the fluticasone propionate CFC inhalation aerosol). Absolute bioavailability of fluticasone propionate was 5.3% and 5.5% following administration of ADVAIR HFA and ADVAIR DISKUS, respectively.
Subjects with Asthma: A double-blind crossover trial was conducted in 13 adult subjects with asthma to evaluate the steady-state pharmacokinetics of fluticasone propionate and salmeterol following administration of 2 inhalations of ADVAIR HFA 115 mcg/21 mcg twice daily or 1 inhalation of ADVAIR DISKUS 250 mcg/50 mcg twice daily for 4 weeks. Systemic exposure (AUC) to fluticasone propionate was similar for ADVAIR HFA (274 pg•h/mL [95% CI: 150, 502]) and ADVAIR DISKUS (338 pg•h/mL [95% CI: 197, 581]).
The effect of 21 days of treatment with ADVAIR HFA 45 mcg/21 mcg (2 inhalations twice daily with or without a spacer) or ADVAIR DISKUS 100 mcg/50 mcg (1 inhalation twice daily) was evaluated in a trial of 31 children aged 4 to 11 years with mild asthma. Systemic exposure to fluticasone propionate was similar with ADVAIR DISKUS and ADVAIR HFA with a spacer (138 pg•h/mL [95% CI: 69, 273] and 107 pg•h/mL [95% CI: 46, 252], respectively) and lower with ADVAIR HFA without a spacer (24 pg•h/mL [95% CI: 10, 60]).
Salmeterol Xinafoate: Healthy Subjects: Salmeterol xinafoate, an ionic salt, dissociates in solution so that the salmeterol and 1-hydroxy-2-naphthoic acid (xinafoate) moieties are absorbed, distributed, metabolized, and eliminated independently. Salmeterol acts locally in the lung; therefore, plasma levels do not predict therapeutic effect.
Peak plasma concentrations of salmeterol (N = 20 subjects) following 8 inhalations of ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, and ADVAIR HFA 230 mcg/21 mcg ranged from 220 to 470 pg/mL.
In 15 healthy subjects receiving ADVAIR HFA 230 mcg/21 mcg (920/84 mcg) and ADVAIR DISKUS 500 mcg/50 mcg (1,000/100 mcg), systemic exposure to salmeterol was higher (317 versus 169 pg•h/mL) and peak salmeterol concentrations were lower (196 versus 223 pg/mL) following ADVAIR HFA compared with ADVAIR DISKUS, although pharmacodynamic results were comparable.
Subjects with Asthma: Because of the small therapeutic dose, systemic levels of salmeterol are low or undetectable after inhalation of recommended dosages (42 mcg of salmeterol inhalation aerosol twice daily). Following chronic administration of an inhaled dose of 42 mcg of salmeterol inhalation aerosol twice daily, salmeterol was detected in plasma within 5 to 10 minutes in 6 subjects with asthma; plasma concentrations were very low, with mean peak concentrations of 150 pg/mL at 20 minutes and no accumulation with repeated doses.
A double-blind crossover trial was conducted in 13 adult subjects with asthma to evaluate the steady-state pharmacokinetics of fluticasone propionate and salmeterol following administration of 2 inhalations of ADVAIR HFA 115 mcg/21 mcg twice daily or 1 inhalation of ADVAIR DISKUS 250 mcg/50 mcg twice daily for 4 weeks. Systemic exposure to salmeterol was similar for ADVAIR HFA (53 pg•h/mL [95% CI: 17, 164]) and ADVAIR DISKUS (70 pg•h/mL [95% CI: 19, 254]).
The effect of 21 days of treatment with ADVAIR HFA 45 mcg/21 mcg (2 inhalations twice daily with or without a spacer) or ADVAIR DISKUS 100 mcg/50 mcg (1 inhalation twice daily) was evaluated in 31 children aged 4 to 11 years with mild asthma. Systemic exposure to salmeterol was similar for ADVAIR HFA, ADVAIR HFA with spacer, and ADVAIR DISKUS (126 pg•h/mL [95% CI: 70, 225], 103 pg•h/mL [95% CI: 54, 200], and 110 pg•h/mL [95% CI: 55, 219], respectively).
Distribution
Fluticasone Propionate: Following intravenous administration, the initial disposition phase for fluticasone propionate was rapid and consistent with its high lipid solubility and tissue binding. The volume of distribution averaged 4.2 L/kg.
The percentage of fluticasone propionate bound to human plasma proteins averages 99%. Fluticasone propionate is weakly and reversibly bound to erythrocytes and is not significantly bound to human transcortin.
Salmeterol: The percentage of salmeterol bound to human plasma proteins averages 96% in vitro over the concentration range of 8 to 7,722 ng of salmeterol base per milliliter, much higher concentrations than those achieved following therapeutic doses of salmeterol.
Elimination
Fluticasone Propionate: Following intravenous dosing, fluticasone propionate showed polyexponential kinetics and had a terminal elimination half-life of approximately 7.8 hours. The total clearance of fluticasone propionate is high (average, 1,093 mL/min), with renal clearance accounting for <0.02% of the total. Less than 5% of a radiolabeled oral dose was excreted in the urine as metabolites, with the remainder excreted in the feces as parent drug and metabolites. Terminal half-life estimates of fluticasone propionate for ADVAIR HFA, ADVAIR DISKUS, and fluticasone propionate CFC inhalation aerosol were similar and averaged 5.6 hours.
Salmeterol: In 2 healthy adult subjects who received 1 mg of radiolabeled salmeterol (as salmeterol xinafoate) orally, approximately 25% and 60% of the radiolabeled salmeterol was eliminated in urine and feces, respectively, over a period of 7 days. The terminal elimination half-life was about 5.5 hours (1 volunteer only).
The xinafoate moiety has no apparent pharmacologic activity. The xinafoate moiety is highly protein bound (>99%) and has a long elimination half-life of 11 days. No terminal half-life estimates were calculated for salmeterol following administration of ADVAIR HFA.
Metabolism: Fluticasone Propionate: The only circulating metabolite detected in man is the 17β-carboxylic acid derivative of fluticasone propionate, which is formed through the CYP3A4 pathway. This metabolite had less affinity (approximately 1/2,000) than the parent drug for the glucocorticoid receptor of human lung cytosol in vitro and negligible pharmacological activity in animal studies. Other metabolites detected in vitro using cultured human hepatoma cells have not been detected in man.
Salmeterol: Salmeterol base is extensively metabolized by hydroxylation, with subsequent elimination predominantly in the feces. No significant amount of unchanged salmeterol base was detected in either urine or feces.
An in vitro study using human liver microsomes showed that salmeterol is extensively metabolized to α-hydroxysalmeterol (aliphatic oxidation) by CYP3A4. Ketoconazole, a strong inhibitor of CYP3A4, essentially completely inhibited the formation of α-hydroxysalmeterol in vitro.
Specific Populations
A population pharmacokinetic analysis was performed for fluticasone propionate and salmeterol utilizing data from 9 controlled clinical trials that included 350 subjects with asthma aged 4 to 77 years who received treatment with ADVAIR DISKUS, ADVAIR HFA, fluticasone propionate inhalation powder (FLOVENT DISKUS), HFA-propelled fluticasone propionate inhalation aerosol (FLOVENT HFA), or CFC-propelled fluticasone propionate inhalation aerosol. The population pharmacokinetic analyses for fluticasone propionate and salmeterol showed no clinically relevant effects of age, gender, race, body weight, body mass index, or percent of predicted FEV 1 on apparent clearance and apparent volume of distribution.
Patients with Hepatic and Renal Impairment: Formal pharmacokinetic studies using ADVAIR HFA have not been conducted in patients with hepatic or renal impairment. However, since both fluticasone propionate and salmeterol are predominantly cleared by hepatic metabolism, impairment of liver function may lead to accumulation of fluticasone propionate and salmeterol in plasma. Therefore, patients with hepatic disease should be closely monitored.
Drug Interaction Studies
In the repeat- and single-dose trials, there was no evidence of significant drug interaction in systemic exposure between fluticasone propionate and salmeterol when given alone or in combination via the DISKUS. Similar definitive studies have not been performed with ADVAIR HFA. The population pharmacokinetic analysis from 9 controlled clinical trials in 350 subjects with asthma showed no significant effects on fluticasone propionate or salmeterol pharmacokinetics following co-administration with beta 2 -agonists, corticosteroids, antihistamines, or theophyllines.
Inhibitors of Cytochrome P450 3A4: Ritonavir: Fluticasone Propionate: Fluticasone propionate is a substrate of CYP3A4. Coadministration of fluticasone propionate and the strong CYP3A4 inhibitor ritonavir is not recommended based upon a multiple-dose, crossover drug interaction trial in 18 healthy subjects. Fluticasone propionate aqueous nasal spray (200 mcg once daily) was coadministered for 7 days with ritonavir (100 mg twice daily). Plasma fluticasone propionate concentrations following fluticasone propionate aqueous nasal spray alone were undetectable (<10 pg/mL) in most subjects, and when concentrations were detectable peak levels (C max ) averaged 11.9 pg/mL (range: 10.8 to 14.1 pg/mL) and AUC (0-τ) averaged 8.43 pg•h/mL (range: 4.2 to 18.8 pg•h/mL). Fluticasone propionate C max and AUC (0-τ) increased to 318 pg/mL (range: 110 to 648 pg/mL) and 3,102.6 pg•h/mL (range: 1,207.1 to 5,662.0 pg•h/mL), respectively, after coadministration of ritonavir with fluticasone propionate aqueous nasal spray. This significant increase in plasma fluticasone propionate exposure resulted in a significant decrease (86%) in serum cortisol AUC.
Ketoconazole: Fluticasone Propionate: In a placebo-controlled crossover trial in 8 healthy adult volunteers, coadministration of a single dose of orally inhaled fluticasone propionate (1,000 mcg) with multiple doses of ketoconazole (200 mg) to steady state resulted in increased plasma fluticasone propionate exposure, a reduction in plasma cortisol AUC, and no effect on urinary excretion of cortisol.
Salmeterol: In a placebo-controlled, crossover drug interaction trial in 20 healthy male and female subjects, coadministration of salmeterol (50 mcg twice daily) and the strong CYP3A4 inhibitor ketoconazole (400 mg once daily) for 7 days resulted in a significant increase in plasma salmeterol exposure as determined by a 16-fold increase in AUC (ratio with and without ketoconazole 15.76 [90% CI: 10.66, 23.31]) mainly due to increased bioavailability of the swallowed portion of the dose. Peak plasma salmeterol concentrations were increased by 1.4-fold (90% CI: 1.23, 1.68). Three (3) out of 20 subjects (15%) were withdrawn from salmeterol and ketoconazole coadministration due to beta-agonist–mediated systemic effects (2 with QTc prolongation and 1 with palpitations and sinus tachycardia). Coadministration of salmeterol and ketoconazole did not result in a clinically significant effect on mean heart rate, mean blood potassium, or mean blood glucose. Although there was no statistical effect on the mean QTc, coadministration of salmeterol and ketoconazole was associated with more frequent increases in QTc duration compared with salmeterol and placebo administration.
Erythromycin: Fluticasone Propionate: In a multiple-dose drug interaction trial, coadministration of orally inhaled fluticasone propionate (500 mcg twice daily) and erythromycin (333 mg 3 times daily) did not affect fluticasone propionate pharmacokinetics.
Salmeterol: In a repeat-dose trial in 13 healthy subjects, concomitant administration of erythromycin (a moderate CYP3A4 inhibitor) and salmeterol inhalation aerosol resulted in a 40% increase in salmeterol C max at steady state (ratio with and without erythromycin 1.4 [90% CI: 0.96, 2.03], P = 0.12), a 3.6-beat/min increase in heart rate ([95% CI: 0.19, 7.03], P <0.04), a 5.8-msec increase in QTc interval ([95% CI: -6.14, 17.77], P = 0.34), and no change in plasma potassium.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Fluticasone Propionate
Fluticasone propionate demonstrated no tumorigenic potential in mice at oral doses up to 1,000 mcg/kg (approximately 5 times the MRHDID on a mcg/m 2 basis) for 78 weeks or in rats at inhalation doses up to 57 mcg/kg (approximately 0.5 times the MRHDID on a mcg/m 2 basis) for 104 weeks.
Fluticasone propionate did not induce gene mutation in prokaryotic or eukaryotic cells in vitro. No significant clastogenic effect was seen in cultured human peripheral lymphocytes in vitro or in the in vivo mouse micronucleus test.
Fertility and reproductive performance were unaffected in male and female rats at subcutaneous doses up to 50 mcg/kg (approximately 0.5 times the MRHDID for adults on a mcg/m 2 basis).
Salmeterol
In an 18-month carcinogenicity study in CD-mice, salmeterol at oral doses of 1,400 mcg/kg and above (approximately 10 times the MRHDID based on comparison of the plasma AUCs) caused a dose-related increase in the incidence of smooth muscle hyperplasia, cystic glandular hyperplasia, leiomyomas of the uterus, and ovarian cysts. No tumors were seen at 200 mcg/kg (approximately 2 times the MRHDID for adults based on comparison of the AUCs).
In a 24-month oral and inhalation carcinogenicity study in Sprague Dawley rats, salmeterol caused a dose-related increase in the incidence of mesovarian leiomyomas and ovarian cysts at doses of 680 mcg/kg and above (approximately 80 times the MRHDID on a mcg/m 2 basis). No tumors were seen at 210 mcg/kg (approximately 25 times the MRHDID on a mcg/m 2 basis). These findings in rodents are similar to those reported previously for other beta-adrenergic agonist drugs. The relevance of these findings to human use is unknown.
Salmeterol produced no detectable or reproducible increases in microbial and mammalian gene mutation in vitro. No clastogenic activity occurred in vitro in human lymphocytes or in vivo in a rat micronucleus test.
Fertility and reproductive performance were unaffected in male and female rats at oral doses up to 2,000 mcg/kg (approximately 230 times the MRHDID for adults on a mcg/m 2 basis).
Animal Toxicology and/or Pharmacology
Studies in laboratory animals (minipigs, rodents, and dogs) have demonstrated the occurrence of cardiac arrhythmias and sudden death (with histologic evidence of myocardial necrosis) when beta-agonists and methylxanthines are administered concurrently. The clinical relevance of these findings is unknown.
CLINICAL STUDIES
ADVAIR HFA has been studied in subjects with asthma aged 12 years and older. ADVAIR HFA has not been studied in subjects younger than 12 years or in subjects with COPD. In clinical trials comparing ADVAIR HFA with its individual components, improvements in most efficacy endpoints were greater with ADVAIR HFA than with the use of either fluticasone propionate or salmeterol alone. In addition, clinical trials showed comparable results between ADVAIR HFA and ADVAIR DISKUS.
Trials Comparing ADVAIR HFA with Fluticasone Propionate Alone or Salmeterol Alone
Four (4) double-blind, parallel-group clinical trials were conducted with ADVAIR HFA in 1,517 adult and adolescent subjects (aged 12 years and older, mean baseline FEV 1 65% to 75% of predicted normal) with asthma that was not optimally controlled on their current therapy. All metered-dose inhaler treatments were inhalation aerosols given as 2 inhalations twice daily, and other maintenance therapies were discontinued.
Trial 1: Clinical Trial with ADVAIR HFA 45 mcg/21 mcg
This placebo-controlled, 12-week, U.S. trial compared ADVAIR HFA 45 mcg/21 mcg with fluticasone propionate CFC inhalation aerosol 44 mcg or salmeterol CFC inhalation aerosol 21 mcg, each given as 2 inhalations twice daily. The primary efficacy endpoints were predose FEV 1 and withdrawals due to worsening asthma. This trial was stratified according to baseline asthma therapy: subjects using beta-agonists (albuterol alone [n = 142], salmeterol [n = 84], or ICS [n = 134] [daily doses of beclomethasone dipropionate 252 to 336 mcg; budesonide 400 to 600 mcg; flunisolide 1,000 mcg; fluticasone propionate inhalation aerosol 176 mcg; fluticasone propionate inhalation powder 200 mcg; or triamcinolone acetonide 600 to 800 mcg]). Baseline FEV 1 measurements were similar across treatments: ADVAIR HFA 45 mcg/21 mcg, 2.29 L; fluticasone propionate 44 mcg, 2.20 L; salmeterol, 2.33 L; and placebo, 2.27 L.
Predefined withdrawal criteria for lack of efficacy, an indicator of worsening asthma, were utilized for this placebo-controlled trial. Worsening asthma was defined as a clinically important decrease in FEV 1 or PEF, increase in use of VENTOLIN (albuterol, USP) Inhalation Aerosol, increase in night awakenings due to asthma, emergency intervention or hospitalization due to asthma, or requirement for asthma medicine not allowed by the protocol. As shown in Table 3, statistically significantly fewer subjects receiving ADVAIR HFA 45 mcg/21 mcg were withdrawn due to worsening asthma compared with salmeterol and placebo. Fewer subjects receiving ADVAIR HFA 45 mcg/21 mcg were withdrawn due to worsening asthma compared with fluticasone propionate 44 mcg; however, the difference was not statistically significant.
ADVAIR HFA 45 mcg/21 mcg (n = 92) | Fluticasone Propionate CFC Inhalation Aerosol 44 mcg (n = 89) | Salmeterol CFC Inhalation Aerosol 21 mcg (n = 92) | Placebo HFA Inhalation Aerosol (n = 87) |
2% | 8% | 25% | 28% |
The FEV 1 results are displayed in Figure 1. Because this trial used predetermined criteria for worsening asthma, which caused more subjects in the placebo group to be withdrawn, FEV 1 results at Endpoint (last available FEV 1 result) are also provided. Subjects receiving ADVAIR HFA 45 mcg/21 mcg had significantly greater improvements in FEV 1 (0.58 L, 27%) compared with fluticasone propionate 44 mcg (0.36 L, 18%), salmeterol (0.25 L, 12%), and placebo (0.14 L, 5%). These improvements in FEV 1 with ADVAIR HFA 45 mcg/21 mcg were achieved regardless of baseline asthma therapy (albuterol alone, salmeterol, or ICS).
Figure 1. Mean Percent Change from Baseline in FEV1 in Subjects Previously Treated with Either Beta 2 ‑agonists (Albuterol or Salmeterol) or Inhaled Corticosteroids (Trial 1)
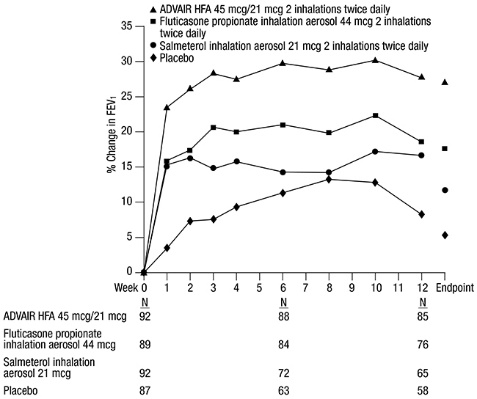
The effect of ADVAIR HFA 45 mcg/21 mcg on the secondary efficacy parameters, including morning and evening PEF, usage of VENTOLIN Inhalation Aerosol, and asthma symptoms over 24 hours on a scale of 0 to 5 is shown in Table 4.
| a Change from baseline = change from baseline at Endpoint (last available data). | ||||
Efficacy Variable a | ADVAIR HFA 45 mcg/21 mcg (n = 92) | Fluticasone Propionate CFC Inhalation Aerosol 44 mcg (n = 89) | Salmeterol CFC Inhalation Aerosol 21 mcg (n = 92) | Placebo HFA Inhalation Aerosol (n = 87) |
AM PEF (L/min) | ||||
| 377 | 369 | 381 | 382 |
| 58 | 27 | 25 | 1 |
PM PEF (L/min) | ||||
| 397 | 387 | 402 | 407 |
| 48 | 20 | 16 | 3 |
Use of VENTOLIN Inhalation Aerosol (inhalations/day) | ||||
| 3.1 | 2.4 | 2.7 | 2.7 |
| -2.1 | -0.4 | -0.8 | 0.2 |
Asthma symptom score/day | ||||
| 1.8 | 1.6 | 1.7 | 1.7 |
| -1.0 | -0.3 | -0.4 | 0 |
The subjective impact of asthma on subjects’ perception of health was evaluated through use of an instrument called the Asthma Quality of Life Questionnaire (AQLQ) (based on a 7-point scale where 1 = maximum impairment and 7 = none). Subjects receiving ADVAIR HFA 45 mcg/21 mcg had clinically meaningful improvements in overall asthma-specific quality of life as defined by a difference between groups of ≥0.5 points in change from baseline AQLQ scores (difference in AQLQ score of 1.14 [95% CI: 0.85, 1.44] compared with placebo).
Trial 2: Clinical Trial with ADVAIR HFA 45 mcg/21 mcg
This active-controlled, 12-week, U.S. trial compared ADVAIR HFA 45 mcg/21 mcg with fluticasone propionate CFC inhalation aerosol 44 mcg and salmeterol CFC inhalation aerosol 21 mcg, each given as 2 inhalations twice daily, in 283 subjects using as-needed albuterol alone. The primary efficacy endpoint was predose FEV 1 . Baseline FEV 1 measurements were similar across treatments: ADVAIR HFA 45 mcg/21 mcg, 2.37 L; fluticasone propionate 44 mcg, 2.31 L; and salmeterol, 2.34 L.
Efficacy results in this trial were similar to those observed in Trial 1. Subjects receiving ADVAIR HFA 45 mcg/21 mcg had significantly greater improvements in FEV 1 (0.69 L, 33%) compared with fluticasone propionate 44 mcg (0.51 L, 25%) and salmeterol (0.47 L, 22%).
Trial 3: Clinical Trial with ADVAIR HFA 115 mcg/21 mcg
This placebo-controlled, 12-week, U.S. trial compared ADVAIR HFA 115 mcg/21 mcg with fluticasone propionate CFC inhalation aerosol 110 mcg or salmeterol CFC inhalation aerosol 21 mcg, each given as 2 inhalations twice daily, in 365 subjects using ICS (daily doses of beclomethasone dipropionate 378 to 840 mcg; budesonide 800 to 1,200 mcg; flunisolide 1,250 to 2,000 mcg; fluticasone propionate inhalation aerosol 440 to 660 mcg; fluticasone propionate inhalation powder 400 to 600 mcg; or triamcinolone acetonide 900 to 1,600 mcg). The primary efficacy endpoints were predose FEV 1 and withdrawals due to worsening asthma. Baseline FEV 1 measurements were similar across treatments: ADVAIR HFA 115 mcg/21 mcg, 2.23 L; fluticasone propionate 110 mcg, 2.18 L; salmeterol, 2.22 L; and placebo, 2.17 L.
Efficacy results in this trial were similar to those observed in Trials 1 and 2. Subjects receiving ADVAIR HFA 115 mcg/21 mcg had significantly greater improvements in FEV 1 (0.41 L, 20%) compared with fluticasone propionate 110 mcg (0.19 L, 9%), salmeterol (0.15 L, 8%), and placebo (-0.12 L, -6%). Significantly fewer subjects receiving ADVAIR HFA 115 mcg/21 mcg were withdrawn from this trial for worsening asthma (7%) compared with salmeterol (24%) and placebo (54%). Fewer subjects receiving ADVAIR HFA 115 mcg/21 mcg were withdrawn due to worsening asthma (7%) compared with fluticasone propionate 110 mcg (11%); however, the difference was not statistically significant.
Trial 4: Clinical Trial with ADVAIR HFA 230 mcg/21 mcg
This active-controlled, 12-week, non-U.S. trial compared ADVAIR HFA 230 mcg/21 mcg with fluticasone propionate CFC inhalation aerosol 220 mcg, each given as 2 inhalations twice daily, and with ADVAIR DISKUS 500 mcg /50 mcg given as 1 inhalation twice daily in 509 subjects using ICS (daily doses of beclomethasone dipropionate CFC inhalation aerosol 1,500 to 2,000 mcg; budesonide 1,500 to 2,000 mcg; flunisolide 1,500 to 2,000 mcg; fluticasone propionate inhalation aerosol 660 to 880 mcg; or fluticasone propionate inhalation powder 750 to 1,000 mcg). The primary efficacy endpoint was morning PEF.
Baseline morning PEF measurements were similar across treatments: ADVAIR HFA 230 mcg/21 mcg, 327 L/min; ADVAIR DISKUS 500 mcg/50 mcg, 341 L/min; and fluticasone propionate 220 mcg, 345 L/min. As shown in Figure 2, morning PEF improved significantly with ADVAIR HFA 230 mcg/21 mcg compared with fluticasone propionate 220 mcg over the 12-week treatment period. Improvements in morning PEF observed with ADVAIR HFA 230 mcg/21 mcg were similar to improvements observed with ADVAIR DISKUS 500 mcg/50 mcg.
Figure 2. Mean Percent Change from Baseline in Morning Peak Expiratory Flow in Subjects Previously Treated with Inhaled Corticosteroids (Trial 4)
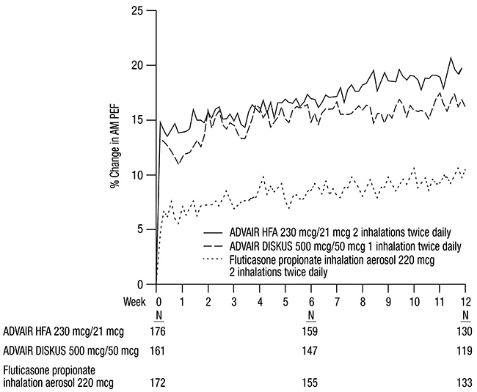
One-Year Safety Trial
Clinical Trial with ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, and ADVAIR HFA 230 mcg/21 mcg
This 1-year, open-label, non-U.S. trial evaluated the safety of ADVAIR HFA 45 mcg/21 mcg, ADVAIR HFA 115 mcg/21 mcg, and ADVAIR HFA 230 mcg/21 mcg given as 2 inhalations twice daily in 325 subjects. This trial was stratified into 3 groups according to baseline asthma therapy: subjects using short-acting beta 2 -agonists alone (n = 42), salmeterol (n = 91), or ICS (n = 277). Subjects treated with short-acting beta 2 -agonists alone, salmeterol, or low doses of ICS with or without concurrent salmeterol received ADVAIR HFA 45 mcg/21 mcg. Subjects treated with moderate doses of ICS with or without concurrent salmeterol received ADVAIR HFA 115 mcg/21 mcg. Subjects treated with high doses of ICS with or without concurrent salmeterol received ADVAIR HFA 230 mcg/21 mcg. Baseline FEV 1 measurements ranged from 2.3 to 2.6 L.
Improvements in FEV 1 (0.17 to 0.35 L at 4 weeks) were seen across all 3 treatments and were sustained throughout the 52-week treatment period. Few subjects (3%) were withdrawn due to worsening asthma over 1 year.
Onset of Action and Progression of Improvement in Control
The onset of action and progression of improvement in asthma control were evaluated in 2 placebo-controlled U.S. trials and 1 active-controlled U.S. trial. Following the first dose, the median time to onset of clinically significant bronchodilatation (≥15% improvement in FEV 1 ) in most subjects was seen within 30 to 60 minutes. Maximum improvement in FEV 1 occurred within 4 hours, and clinically significant improvement was maintained for 12 hours (Figure 3).
Following the initial dose, predose FEV 1 relative to Day 1 baseline improved markedly over the first week of treatment and continued to improve over the 12 weeks of treatment in all 3 trials.
No diminution in the 12-hour bronchodilator effect was observed with either ADVAIR HFA 45 mcg/21 mcg (Figures 3 and 4) or ADVAIR HFA 230/21 as assessed by FEV 1 following 12 weeks of therapy.
Figure 3. Percent Change in Serial 12-Hour FEV1 in Subjects Previously Using Either Beta 2 -agonists (Albuterol or Salmeterol) or Inhaled Corticosteroids (Trial 1)
First Treatment Day
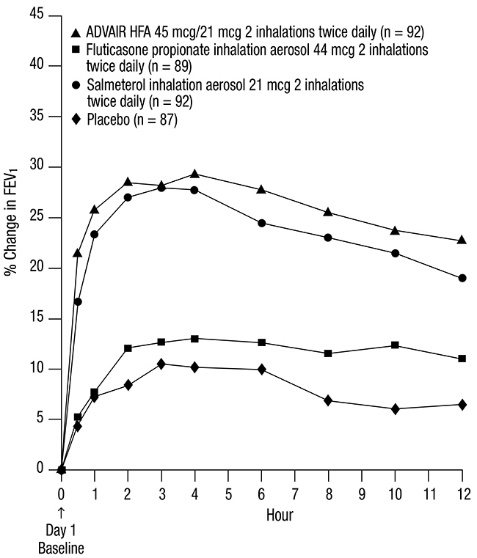
Last Treatment Day (Week 12)
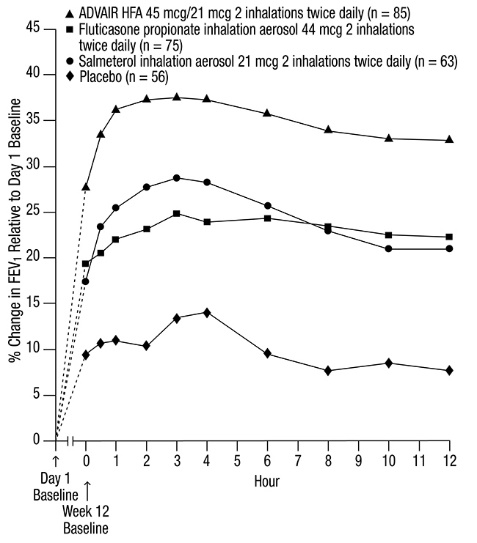
Reduction in asthma symptoms and use of rescue VENTOLIN Inhalation Aerosol and improvement in morning and evening PEF also occurred within the first day of treatment with ADVAIR HFA and continued to improve over the 12 weeks of therapy in all 3 trials.
HOW SUPPLIED/STORAGE AND HANDLING
ADVAIR HFA is supplied in the following boxes of 1 as a pressurized aluminum canister fitted with a counter and supplied with a purple plastic actuator with a light purple cap:
- ADVAIR HFA 45 mcg/21 mcg: 12-g canister containing 120 actuations (NDC 0173-0715-20)
- ADVAIR HFA 45 mcg/21 mcg: 8-g canister containing 60 actuations (NDC 0173-0715-22)
- ADVAIR HFA 115 mcg/21 mcg: 12-g canister containing 120 actuations (NDC 0173-0716-20)
- ADVAIR HFA 115 mcg/21 mcg: 8-g canister containing 60 actuations (NDC 0173-0716-22)
- ADVAIR HFA 230 mcg/21 mcg: 12-g canister containing 120 actuations (NDC 0173-0717-20)
- ADVAIR HFA 230 mcg/21 mcg: 8-g canister containing 60 actuations (NDC 0173-0717-22)
Each inhaler is packaged with a Patient Information leaflet.
The purple actuator supplied with ADVAIR HFA should not be used with any other product canisters, and actuators from other products should not be used with an ADVAIR HFA canister.
Counter
ADVAIR HFA has a counter attached to the canister. The counter starts at 124 or 064 and counts down each time a spray is released. The correct amount of medication in each actuation cannot be assured after the counter reads 000, even though the canister is not completely empty and will continue to operate. The inhaler should be discarded when the counter reads 000.
Contents under Pressure
Do not puncture. Do not use or store near heat or open flame. Exposure to temperatures above 120°F may cause bursting. Never throw canister into fire or incinerator.
Storage
Store at room temperature between 68°F and 77°F (20°C and 25°C); excursions permitted from 59°F to 86°F (15°C to 30°C) [See USP Controlled Room Temperature]. Store the inhaler with the mouthpiece down. For best results, the inhaler should be at room temperature before use.
INSTRUCTIONS FOR USE ADVAIR (AD vair) HFA (fluticasone propionate and salmeterol inhalation aerosol) for oral inhalation use | |
Your ADVAIR HFA inhaler | |
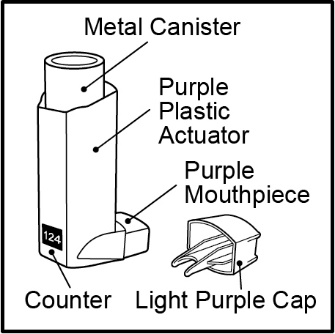 Figure A |
|
Before using your ADVAIR HFA inhaler
Priming your ADVAIR HFA inhaler | |
 Figure B 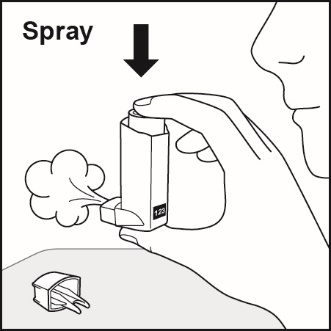 Figure C  Figure D |
|
How to use your ADVAIR HFA inhaler Follow these steps every time you use ADVAIR HFA. | |
 Figure E  Figure F 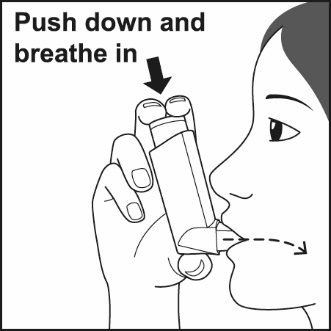 Figure G 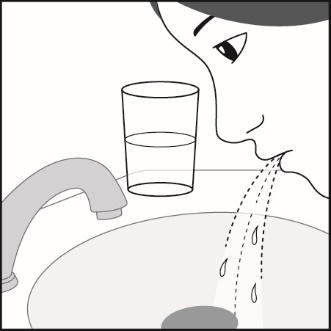 Figure H |
|
| |
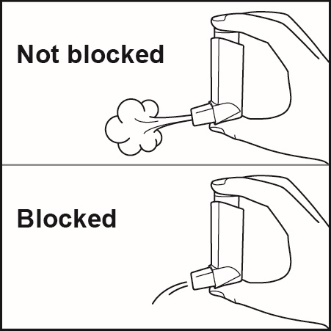 Figure I  Figure J | Clean your inhaler at least 1 time each week after your evening dose. You may not see any medicine build-up on the inhaler, but it is important to keep it clean so medicine build-up will not block the spray. See Figure I.
|
Replacing your ADVAIR HFA inhaler
For correct use of your ADVAIR HFA inhaler, remember:
For more information about ADVAIR HFA or how to use your inhaler, call 1-888-825-5249. Trademarks are owned by or licensed to the GSK group of companies. GlaxoSmithKline, Durham, NC 27701 ©2023 GSK group of companies or its licensor. ADH:7IFU | |
| |
Mechanism of Action
ADVAIR HFA
ADVAIR HFA contains both fluticasone propionate and salmeterol. The mechanisms of action described below for the individual components apply to ADVAIR HFA. These drugs represent 2 different classes of medications (a synthetic corticosteroid and a LABA) that have different effects on clinical, physiologic, and inflammatory indices of asthma.
Fluticasone Propionate
Fluticasone propionate is a synthetic trifluorinated corticosteroid with anti-inflammatory activity. Fluticasone propionate has been shown in vitro to exhibit a binding affinity for the human glucocorticoid receptor that is 18 times that of dexamethasone, almost twice that of beclomethasone-17-monopropionate (BMP), the active metabolite of beclomethasone dipropionate, and over 3 times that of budesonide. Data from the McKenzie vasoconstrictor assay in man are consistent with these results. The clinical significance of these findings is unknown.
Inflammation is an important component in the pathogenesis of asthma. Corticosteroids have been shown to have a wide range of actions on multiple cell types (e.g., mast cells, eosinophils, neutrophils, macrophages, lymphocytes) and mediators (e.g., histamine, eicosanoids, leukotrienes, cytokines) involved in inflammation. These anti-inflammatory actions of corticosteroids contribute to their efficacy in asthma.
Salmeterol Xinafoate
Salmeterol is a selective LABA. In vitro studies show salmeterol to be at least 50 times more selective for beta 2 ‑adrenoceptors than albuterol. Although beta 2 -adrenoceptors are the predominant adrenergic receptors in bronchial smooth muscle and beta 1 -adrenoceptors are the predominant receptors in the heart, there are also beta 2 -adrenoceptors in the human heart comprising 10% to 50% of the total beta-adrenoceptors. The precise function of these receptors has not been established, but their presence raises the possibility that even selective beta 2 -agonists may have cardiac effects.
The pharmacologic effects of beta 2 -adrenoceptor agonist drugs, including salmeterol, are at least in part attributable to stimulation of intracellular adenyl cyclase, the enzyme that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3′,5′-adenosine monophosphate (cyclic AMP). Increased cyclic AMP levels cause relaxation of bronchial smooth muscle and inhibition of release of mediators of immediate hypersensitivity from cells, especially from mast cells.
In vitro tests show that salmeterol is a potent and long-lasting inhibitor of the release of mast cell mediators, such as histamine, leukotrienes, and prostaglandin D 2 , from human lung. Salmeterol inhibits histamine-induced plasma protein extravasation and inhibits platelet-activating factor–induced eosinophil accumulation in the lungs of guinea pigs when administered by the inhaled route. In humans, single doses of salmeterol administered via inhalation aerosol attenuate allergen-induced bronchial hyper-responsiveness.