Get your patient on Emgality (Galcanezumab-Gnlm)
Patient education
Patient education materials
Dosing resources
Clinical information
Insurance resources
Financial assistance & copay programs
Other resources
Dosage & administration

Emgality prescribing information
| Warnings and Precautions (5.2 ) | 06/2026 |
INDICATIONS AND USAGE
EMGALITY ® is a calcitonin-gene related peptide antagonist indicated in adults for the:
Migraine
EMGALITY is indicated for the preventive treatment of migraine in adults.
Episodic Cluster Headache
EMGALITY is indicated for the treatment of episodic cluster headache in adults.
DOSAGE AND ADMINISTRATION
- For subcutaneous use only. (2.1 , 2.2 , 2.3 )
- Migraine recommended dosage: 240 mg loading dose (administered as two consecutive injections of 120 mg each), followed by monthly doses of 120 mg. (2.1 )
- Episodic cluster headache recommended dosage: 300 mg (administered as three consecutive injections of 100 mg each) at the onset of the cluster period, and then monthly until the end of the cluster period. (2.2 )
- Administer in the abdomen, thigh, back of the upper arm, or buttocks subcutaneously. (2.3 )
Recommended Dosing for Migraine
The recommended dosage of EMGALITY is 240 mg (two consecutive subcutaneous injections of 120 mg each) once as a loading dose, followed by monthly doses of 120 mg injected subcutaneously.
If a dose of EMGALITY is missed, administer as soon as possible. Thereafter, EMGALITY can be scheduled monthly from the date of the last dose.
Recommended Dosing for Episodic Cluster Headache
The recommended dosage of EMGALITY is 300 mg (three consecutive subcutaneous injections of 100 mg each) at the onset of the cluster period, and then monthly until the end of the cluster period.
If a dose of EMGALITY is missed during a cluster period, administer as soon as possible. Thereafter, EMGALITY can be scheduled monthly from the date of the last dose until the end of the cluster period.
Important Administration Instructions
EMGALITY is for subcutaneous use only.
EMGALITY is intended for patient self-administration. Prior to use, provide proper training to patients and/or caregivers on how to prepare and administer EMGALITY using the single-dose prefilled pen or single-dose prefilled syringe, including aseptic technique [see How Supplied/Storage and Handling (16.2 ) and Instructions for Use] :
- Protect EMGALITY from direct sunlight.
- Prior to subcutaneous administration, allow EMGALITY to sit at room temperature for 30 minutes. Do not warm by using a heat source such as hot water or a microwave.
- Do not shake the product.
- Inspect EMGALITY visually for particulate matter and discoloration prior to administration, whenever solution and container permit [see Dosage Forms and Strengths (3 ) and How Supplied/Storage and Handling (16.1 )] . Do not use EMGALITY if it is cloudy or there are visible particles.
- Administer EMGALITY in the abdomen, thigh, back of the upper arm, or buttocks subcutaneously. Do not inject into areas where the skin is tender, bruised, red, or hard.
- Both the prefilled pen and prefilled syringe are single-dose and deliver the entire contents.
DOSAGE FORMS AND STRENGTHS
EMGALITY is a sterile clear to opalescent, colorless to slightly yellow to slightly brown solution available as follows:
- Injection: 120 mg/mL in a single-dose prefilled pen
- Injection: 120 mg/mL in a single-dose prefilled syringe
- Injection: 100 mg/mL in a single-dose prefilled syringe
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to EMGALITY during pregnancy. Healthcare providers are encouraged to register pregnant patients, or pregnant women may enroll themselves in the registry by calling 1-833-464-4724 or by contacting the company at www.migrainepregnancyregistry.com.
Risk Summary
There are no adequate data on the developmental risk associated with the use of EMGALITY in pregnant women. Administration of galcanezumab-gnlm to rats and rabbits during the period of organogenesis or to rats throughout pregnancy and lactation at plasma exposures greater than that expected clinically did not result in adverse effects on development (see Animal Data) .
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% - 4% and 15% - 20%, respectively. The estimated rate of major birth defects (2.2% - 2.9%) and miscarriage (17%) among deliveries to women with migraine are similar to rates reported in women without migraine.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data have suggested that women with migraine may be at increased risk of preeclampsia during pregnancy.
Data
Animal Data
When galcanezumab-gnlm was administered to female rats by subcutaneous injection in two studies (0, 30, or 100 mg/kg; 0 or 250 mg/kg) prior to and during mating and continuing throughout organogenesis, no adverse effects on embryofetal development were observed. The highest dose tested (250 mg/kg) was associated with a plasma exposure (C ave, ss ) 38 or 18 times that in humans at the recommended human dose (RHD) for migraine (120 mg) or episodic cluster headache (300 mg), respectively. Administration of galcanezumab-gnlm (0, 30, or 100 mg/kg) by subcutaneous injection to pregnant rabbits throughout the period of organogenesis produced no adverse effects on embryofetal development. The higher dose tested was associated with a plasma C ave, ss 64 or 29 times that in humans at 120 mg or 300 mg, respectively.
Administration of galcanezumab-gnlm (0, 30, or 250 mg/kg) by subcutaneous injection to rats throughout pregnancy and lactation produced no adverse effects on pre- and postnatal development. The higher dose tested was associated with a plasma C ave, ss 34 or 16 times that in humans at 120 mg or 300 mg, respectively.
Lactation
Risk Summary
There are no data on the presence of galcanezumab-gnlm in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for EMGALITY and any potential adverse effects on the breastfed infant from EMGALITY or from the underlying maternal condition.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Clinical studies of EMGALITY did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.
CONTRAINDICATIONS
EMGALITY is contraindicated in patients with serious hypersensitivity to galcanezumab-gnlm or to any of the excipients [see Warnings and Precautions (5.1 )] .
WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions: If a serious hypersensitivity reaction occurs, discontinue administration of EMGALITY and initiate appropriate therapy. Hypersensitivity reactions can occur days after administration, and may be prolonged. (5.1 )
- Constipation with Serious Complications: Serious complications of constipation may occur. (5.2 )
- Hypertension: New-onset or worsening of pre-existing hypertension may occur. (5.3 )
- Raynaud's Phenomenon: New-onset or worsening of pre-existing Raynaud's phenomenon may occur. (5.4 )
Hypersensitivity Reactions
Hypersensitivity reactions, including dyspnea, urticaria, and rash, have occurred with EMGALITY in clinical studies and the postmarketing setting. Cases of anaphylaxis and angioedema have also been reported in the postmarketing setting. If a serious or severe hypersensitivity reaction occurs, discontinue administration of EMGALITY and initiate appropriate therapy [see Contraindications (4 ), Adverse Reactions (6.1 ), and Patient Counseling Information (17 )] . Hypersensitivity reactions can occur days after administration and may be prolonged.
Constipation with Serious Complications
Constipation with serious complications has been reported following the use of monoclonal antibody CGRP antagonists, including EMGALITY, in the postmarketing setting. There were cases with monoclonal antibody CGRP antagonists that required hospitalization, including cases where surgery was necessary. In a majority of these cases, the onset of constipation was reported after the first dose; however, patients have also presented with constipation later on in treatment. The monoclonal antibody CGRP antagonist was discontinued in many of the reported cases of constipation with serious complications.
Monitor patients treated with EMGALITY for severe constipation and manage as clinically appropriate. The concurrent use of medications that reduce gastrointestinal motility may increase the risk for more severe constipation and the potential for constipation-related complications.
Hypertension
Development of hypertension and worsening of pre-existing hypertension have been reported following the use of CGRP antagonists, including EMGALITY, in the postmarketing setting. Some of the patients who developed new-onset hypertension had risk factors for hypertension. There were cases requiring initiation of pharmacological treatment for hypertension and, in some cases, hospitalization. Hypertension may occur at any time during treatment but was most frequently reported within 7 days of therapy initiation. EMGALITY was discontinued in many of the reported cases.
Monitor patients treated with EMGALITY for new-onset hypertension or worsening of pre-existing hypertension, and consider whether discontinuation of EMGALITY is warranted if evaluation fails to establish an alternative etiology or blood pressure is inadequately controlled.
Raynaud's Phenomenon
Development of Raynaud’s phenomenon and recurrence or worsening of pre-existing Raynaud’s phenomenon have been reported in the postmarketing setting following the use of CGRP antagonists, including EMGALITY. In reported cases with monoclonal antibody CGRP antagonists, symptom onset occurred after a median of 71 days following dosing. Many of the cases reported serious outcomes, including hospitalizations and disability, generally related to debilitating pain. In most reported cases, discontinuation of the CGRP antagonist resulted in resolution of symptoms.
EMGALITY should be discontinued if signs or symptoms of Raynaud’s phenomenon develop, and patients should be evaluated by a healthcare provider if symptoms do not resolve. Patients with a history of Raynaud’s phenomenon should be monitored for, and informed about the possibility of, worsening or recurrence of signs and symptoms.
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in clinical trials of another drug and may not reflect the rates observed in clinical practice.
Migraine
The safety of EMGALITY has been evaluated in 2586 patients with migraine who received at least one dose of EMGALITY, representing 1487 patient-years of exposure. Of these, 1920 patients were exposed to EMGALITY once monthly for at least 6 months, and 526 patients were exposed for 12 months.
In placebo-controlled clinical studies (Studies 1, 2, and 3), 705 patients received at least one dose of EMGALITY 120 mg once monthly, and 1451 patients received placebo, during 3 months or 6 months of double-blind treatment [see Clinical Studies (14.1 )] . Of the EMGALITY-treated patients, approximately 85% were female, 77% were white, and the mean age was 41 years at study entry.
The most common adverse reaction was injection site reactions. In Studies 1, 2, and 3, 1.8% of patients discontinued double-blind treatment because of adverse events. Table 1 summarizes the adverse reactions that occurred within up to 6 months of treatment in the migraine studies.
a Injection site reactions include multiple related adverse event terms, such as injection site pain, injection site reaction, injection site erythema, and injection site pruritus. | ||
| Adverse Reaction | EMGALITY 120 mg Monthly (N=705) % | Placebo Monthly (N=1451) % |
| Injection site reactions a | 18 | 13 |
Episodic Cluster Headache
EMGALITY was studied for up to 2 months in a placebo-controlled trial in patients with episodic cluster headache (Study 4) [see Clinical Studies (14.2 )] . A total of 106 patients were studied (49 on EMGALITY and 57 on placebo). Of the EMGALITY-treated patients, approximately 84% were male, 88% were white, and the mean age was 47 years at study entry. Two EMGALITY-treated patients discontinued double-blind treatment because of adverse events.
Overall, the safety profile observed in patients with episodic cluster headache treated with EMGALITY 300 mg monthly is consistent with the safety profile in migraine patients.
Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease.
For these reasons, comparison of the incidence of antibodies to galcanezumab-gnlm in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
The immunogenicity of EMGALITY has been evaluated using an in vitro immunoassay for the detection of binding anti-galcanezumab-gnlm antibodies. For patients whose sera tested positive in the screening immunoassay, an in vitro ligand-binding immunoassay was performed to detect neutralizing antibodies.
In controlled studies with EMGALITY up to 6 months (Study 1, Study 2, and Study 3), the incidence of anti-galcanezumab-gnlm antibody development was 4.8% (33/688) in patients receiving EMGALITY once monthly (32 out of 33 of whom had in vitro neutralizing activity). With 12 months of treatment in an open-label study, up to 12.5% (16/128) of EMGALITY-treated patients developed anti-galcanezumab-gnlm antibodies, most of whom tested positive for neutralizing antibodies.
Although anti-galcanezumab-gnlm antibody development was not found to affect the pharmacokinetics, safety or efficacy of EMGALITY in these patients, the available data are too limited to make definitive conclusions.
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of EMGALITY. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to EMGALITY exposure.
Gastrointestinal Disorders — Constipation [see Warnings and Precautions (5.2 )]
Immune System Disorders — Anaphylaxis, angioedema [see Contraindications (4 ) and Warnings and Precautions (5.1 )]
Skin and Subcutaneous Tissue Disorders — Rash
Vascular Disorders — Hypertension [see Warnings and Precautions (5.3 )] , Raynaud's Phenomenon [see Warnings and Precautions (5.4 )]
DESCRIPTION
Galcanezumab-gnlm is a humanized IgG4 monoclonal antibody specific for calcitonin-gene related peptide (CGRP) ligand. Galcanezumab-gnlm is produced in Chinese Hamster Ovary (CHO) cells by recombinant DNA technology. Galcanezumab-gnlm is composed of two identical immunoglobulin kappa light chains and two identical immunoglobulin gamma heavy chains and has an overall molecular weight of approximately 147 kDa.
EMGALITY (galcanezumab-gnlm) injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution, for subcutaneous use. EMGALITY is supplied in a 1 mL single-dose prefilled pen to deliver 120 mg of galcanezumab-gnlm or a 1 mL single-dose prefilled syringe to deliver 100 mg or 120 mg of galcanezumab-gnlm. Each mL of solution contains 100 mg or 120 mg of galcanezumab-gnlm; L-histidine (0.5 mg); L-histidine hydrochloride monohydrate (1.5 mg); Polysorbate 80 (0.5 mg); Sodium Chloride (8.8 mg); Water for Injection, USP. The pH range is 5.3 - 6.3.
CLINICAL PHARMACOLOGY
Mechanism of Action
Galcanezumab-gnlm is a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP) ligand and blocks its binding to the receptor.
Pharmacodynamics
There are no relevant data on the pharmacodynamic effects of galcanezumab-gnlm.
Pharmacokinetics
Galcanezumab-gnlm exhibits linear pharmacokinetics and exposure increases proportionally with doses between 1 and 600 mg.
A loading dose of 240 mg achieved the serum galcanezumab-gnlm steady-state concentration after the first dose. A dose of 300 mg monthly would achieve steady-state concentration after the fourth dose. The time to maximum concentration is 5 days, and the elimination half-life is 27 days.
There was no difference in pharmacokinetic parameters between healthy volunteers, patients with episodic or chronic migraine, and patients with episodic cluster headache.
Absorption
Following a subcutaneous dose of galcanezumab-gnlm, the time to maximum concentration was about 5 days.
Injection site location did not significantly influence the absorption of galcanezumab-gnlm.
Distribution
The apparent volume of distribution (V/F) of galcanezumab-gnlm was 7.3 L (34% Inter Individual Variability [IIV]).
Metabolism and Elimination
Galcanezumab-gnlm is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
The apparent clearance (CL/F) of galcanezumab-gnlm was 0.008 L/h and the elimination half-life of galcanezumab was approximately 27 days.
Specific Populations
Age, Sex, Weight, Race, Ethnicity
The pharmacokinetics of galcanezumab-gnlm were not affected by age, sex, race, subtypes of migraine spectrum (episodic or chronic migraine), or headache diagnosis (migraine vs. episodic cluster headache) based on a population pharmacokinetics analysis. Body weight has no clinically relevant effect on the pharmacokinetics of galcanezumab-gnlm.
Patients with Renal or Hepatic Impairment
Renal and hepatic impairment are not expected to affect the pharmacokinetics of galcanezumab-gnlm. Population pharmacokinetic analysis of integrated data from the galcanezumab-gnlm clinical studies revealed that creatinine clearance did not affect the pharmacokinetics of galcanezumab-gnlm in patients with mild or moderate renal impairment. Patients with severe renal impairment (creatinine clearance <30 mL/min) have not been studied. Based on a population PK analysis, bilirubin concentration did not significantly influence the CL/F of galcanezumab-gnlm.
No dedicated clinical studies were conducted to evaluate the effect of hepatic impairment or renal impairment on the pharmacokinetics of galcanezumab-gnlm.
Drug Interaction Studies
P450 Enzymes
Galcanezumab-gnlm is not metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of galcanezumab-gnlm has not been assessed.
Mutagenesis
Genetic toxicology studies of galcanezumab-gnlm have not been conducted.
Impairment of Fertility
When galcanezumab-gnlm (0, 30, or 250 mg/kg) was administered to male rats by subcutaneous injection prior to and during mating, no adverse effects on fertility was observed. The higher dose tested was associated with a plasma exposure (C ave, ss ) 8 or 4 times that in humans at the recommended human dose (RHD) for migraine (120 mg) or episodic cluster headache (300 mg), respectively. When galcanezumab-gnlm was administered to female rats by subcutaneous injection in two studies (0, 30, or 100 mg/kg; 0 or 250 mg/kg) prior to and during mating and continuing throughout organogenesis, no adverse effects on fertility were observed. The highest dose tested (250 mg/kg) was associated with a plasma C ave, ss 38 or 18 times that in humans at 120 mg or 300 mg, respectively.
CLINICAL STUDIES
Migraine
The efficacy of EMGALITY was evaluated as a preventive treatment of episodic or chronic migraine in three multicenter, randomized, double-blind, placebo-controlled studies: two 6-month studies in patients with episodic migraine (Studies 1 and 2) and one 3-month study in patients with chronic migraine (Study 3).
Episodic Migraine
Study 1 (NCT02614183) and Study 2 (NCT02614196) included adults with a history of episodic migraine (4 to 14 migraine days per month). All patients were randomized in a 1:1:2 ratio to receive once-monthly subcutaneous injections of EMGALITY 120 mg, EMGALITY 240 mg, or placebo. All patients in the 120 mg EMGALITY group received an initial 240 mg loading dose. Patients were allowed to use acute headache treatments, including migraine-specific medications (i.e., triptans, ergotamine derivatives), NSAIDs, and acetaminophen during the study.
The studies excluded patients on any other migraine preventive treatment, patients with medication overuse headache, patients with ECG abnormalities compatible with an acute cardiovascular event and patients with a history of stroke, myocardial infarction, unstable angina, percutaneous coronary intervention, coronary artery bypass grafting, deep vein thrombosis, or pulmonary embolism within 6 months of screening.
The primary efficacy endpoint for Studies 1 and 2 was the mean change from baseline in the number of monthly migraine headache days over the 6-month treatment period. Key secondary endpoints included response rates (the mean percentages of patients reaching at least 50%, 75%, and 100% reduction from baseline in the number of monthly migraine headache days over the 6-month treatment period), the mean change from baseline in the number of monthly migraine headache days with use of any acute headache medication during the 6-month treatment period, and the impact of migraine on daily activities, as assessed by the mean change from baseline in the average Migraine-Specific Quality of Life Questionnaire version 2.1 (MSQ v2.1) Role Function-Restrictive domain score during the last 3 months of treatment (Months 4 to 6). Scores are scaled from 0 to 100, with higher scores indicating less impact of migraine on daily activities.
In Study 1, a total of 858 patients (718 females, 140 males) ranging in age from 18 to 65 years, were randomized. A total of 703 patients completed the 6-month double-blind phase. In Study 2, a total of 915 patients (781 female, 134 male) ranging in age from 18 to 65 years, were randomized. A total of 785 patients completed the 6-month double-blind phase. In Study 1 and Study 2, the mean migraine frequency at baseline was approximately 9 migraine days per month, and was similar across treatment groups.
EMGALITY 120 mg demonstrated statistically significant improvements for efficacy endpoints compared to placebo over the 6-month period, as summarized in Table 2 . EMGALITY treatment with the 240 mg once-monthly dose showed no additional benefit over the EMGALITY 120 mg once-monthly dose.
a p<0.001 | ||||
b N = 189 for EMGALITY 120 mg and N = 377 for placebo in Study 1; N = 213 for EMGALITY 120 mg and N = 396 for placebo in Study 2. | ||||
| Study 1 | Study 2 | |||
| EMGALITY 120 mg N = 210 | Placebo N = 425 | EMGALITY 120 mg N = 226 | Placebo N = 450 | |
| Monthly Migraine Headache Days (over Months 1 to 6) | ||||
| Baseline migraine headache days | 9.2 | 9.1 | 9.1 | 9.2 |
| Mean change from baseline | -4.7 | -2.8 | -4.3 | -2.3 |
| Difference from placebo a | -1.9 | -2.0 | ||
| ≥50% Migraine Headache Days Responders (over Months 1 to 6) | ||||
| % Responders a | 62% | 39% | 59% | 36% |
| ≥75% Migraine Headache Days Responders (over Months 1 to 6) | ||||
| % Responders a | 39% | 19% | 34% | 18% |
| 100% Migraine Headache Days Responders (over Months 1 to 6) | ||||
| % Responders a | 16% | 6% | 12% | 6% |
| Monthly Migraine Headache Days that Acute Medication was Taken (over Months 1 to 6) | ||||
| Mean change from baseline (days) a | -4.0 | -2.2 | -3.7 | -1.9 |
| MSQ Role Function-Restrictive Domain Score (over Months 4 to 6) | ||||
| Baseline | 51.4 | 52.9 | 52.5 | 51.4 |
| Mean change from baseline b | 32.4 | 24.7 | 28.5 | 19.7 |
| Difference from placebo a | 7.7 | 8.8 | ||
Figure 1: Change from Baseline in Monthly Migraine Headache Days in Study 1 a

a Least-square means and 95% confidence intervals are presented.
Figure 2: Change from Baseline in Monthly Migraine Headache Days in Study 2 a

a Least-square means and 95% confidence intervals are presented.
Figure 3 shows the distribution of change from baseline in the mean number of monthly migraine headache days in bins of 2 days, by treatment group, in Study 1. A treatment benefit over placebo for EMGALITY is seen across a range of changes from baseline in monthly migraine headache days.
Figure 3: Distribution of Change from Baseline in Mean Monthly Migraine Headache Days over Months 1 to 6 by Treatment Group in Study 1

Figure 4 shows the distribution of change from baseline in the mean number of monthly migraine headache days in bins of 2 days, by treatment group, in Study 2. A treatment benefit over placebo for EMGALITY is seen across a range of changes from baseline in monthly migraine headache days.
Figure 4: Distribution of Change from Baseline in Mean Monthly Migraine Headache Days over Months 1 to 6 by Treatment Group in Study 2

Chronic Migraine
Study 3 (NCT02614261) included adults with a history of chronic migraine (≥15 headache days per month with ≥8 migraine days per month). All patients were randomized in a 1:1:2 ratio to receive once-monthly subcutaneous injections of EMGALITY 120 mg, EMGALITY 240 mg, or placebo over a 3-month treatment period. All patients in the 120 mg EMGALITY group received an initial 240 mg loading dose.
Patients were allowed to use acute headache treatments including migraine-specific medications (i.e., triptans, ergotamine derivatives), NSAIDs, and acetaminophen. A subset of patients (15%) was allowed to use one concomitant migraine preventive medication. Patients with medication overuse headache were allowed to enroll.
The study excluded patients with ECG abnormalities compatible with an acute cardiovascular event, and patients with a history of stroke, myocardial infarction, unstable angina, percutaneous coronary intervention, coronary artery bypass grafting, deep vein thrombosis, or pulmonary embolism within 6 months of screening.
The primary endpoint was the mean change from baseline in the number of monthly migraine headache days over the 3-month treatment period. The secondary endpoints were response rates (the mean percentages of patients reaching at least 50%, 75% and 100% reduction from baseline in the number of monthly migraine headache days over the 3-month treatment period), the mean change from baseline in the number of monthly migraine headache days with use of any acute headache medication during the 3-month treatment period, and the impact of migraine on daily activities as assessed by the mean change from baseline in the MSQ v2.1 Role Function-Restrictive domain score at Month 3. Scores are scaled from 0 to 100, with higher scores indicating less impact of migraine on daily activities.
In Study 3, a total of 1113 patients (946 female, 167 male) ranging in age from 18 to 65 years, were randomized. A total of 1037 patients completed the 3-month double-blind phase. The mean number of monthly migraine headache days at baseline was approximately 19.
EMGALITY 120 mg demonstrated statistically significant improvement for the mean change from baseline in the number of monthly migraine headache days over the 3-month treatment period, and in the mean percentage of patients reaching at least 50% reduction from baseline in the number of monthly migraine headache days over the 3-month treatment period, as summarized in Table 3 . EMGALITY treatment with the 240 mg once-monthly dose showed no additional benefit over the EMGALITY 120 mg once-monthly dose.
a p<0.001 | ||
| EMGALITY 120 mg N = 273 | Placebo N =538 | |
| Monthly Migraine Headache Days (over Months 1 to 3) | ||
| Baseline migraine headache days | 19.4 | 19.6 |
| Mean change from baseline | -4.8 | -2.7 |
| Difference from placebo a | -2.1 | |
| ≥50% Migraine Headache Days Responders (over Months 1 to 3) | ||
| % Responders a | 28% | 15% |
Study 3 utilized a sequential testing procedure to control the Type-I error rate for the multiple secondary endpoints. Once a secondary endpoint failed to reach the required level for statistical significance, formal hypothesis testing was terminated for subsequent endpoints, and p-values were considered nominal only. In Study 3, EMGALITY 120 mg was not significantly better than placebo for the proportion of patients with ≥75% or 100% reduction in migraine headache days. Patients treated with EMGALITY 120 mg showed a nominally greater reduction in the number of monthly migraine headache days that acute medication was taken (-4.7 for EMGALITY 120 mg vs. -2.2 for placebo; nominal p-value <0.001), and the mean change from baseline in the MSQ Role Function-Restrictive Domain score at Month 3 was nominally greater in patients treated with EMGALITY 120 mg than in patients on placebo (21.8 for EMGALITY 120 mg vs. 16.8 for placebo; nominal p-value <0.001).
Figure 5: Change from Baseline in Monthly Migraine Headache Days in Study 3 a

a Least-square means and 95% confidence intervals are presented.
Figure 6 shows the distribution of change from baseline in the mean number of monthly migraine headache days for the 3-month study period in bins of 3 days by treatment group. A treatment benefit over placebo for EMGALITY is seen across a range of changes from baseline in monthly migraine headache days.
Figure 6: Distribution of Change from Baseline in Mean Monthly Migraine Headache Days over Months 1 to 3 by Treatment Group in Study 3

Episodic Cluster Headache
The efficacy of EMGALITY was evaluated for the treatment of episodic cluster headache in a randomized, 8-week, double-blind, placebo-controlled study (Study 4).
Study 4 (NCT02397473) included adults who met the International Classification of Headache Disorders 3 rd edition (beta version) diagnostic criteria for episodic cluster headache and had a maximum of 8 attacks per day, a minimum of one attack every other day, and at least 4 attacks during the prospective 7-day baseline period. All patients were randomized in a 1:1 ratio to receive once-monthly subcutaneous injections of EMGALITY 300 mg or placebo. Patients were allowed to use certain specified acute/abortive cluster headache treatments, including triptans, oxygen, acetaminophen, and NSAIDs during the study.
The study excluded patients on other treatments intended to reduce the frequency of cluster headache attacks; patients with medication overuse headache; patients with ECG abnormalities compatible with an acute cardiovascular event or conduction delay; and patients with a history of myocardial infarction, unstable angina, percutaneous coronary intervention, coronary artery bypass grafting, deep vein thrombosis, or pulmonary embolism within 6 months of screening. In addition, patients with any history of stroke, intracranial or carotid aneurysm, intracranial hemorrhage, or vasospastic angina; clinical evidence of peripheral vascular disease; or diagnosis of Raynaud’s disease were excluded.
The primary efficacy endpoint for Study 4 was the mean change from baseline in weekly cluster headache attack frequency across Weeks 1 to 3. A secondary endpoint was the percentage of patients who achieved a response (defined as a reduction from baseline of 50% or greater in the weekly cluster headache attack frequency) at Week 3.
In Study 4, a total of 106 patients (88 males, 18 females) ranging in age from 19 to 65 years were randomized and treated. A total of 90 patients completed the 8-week double-blind phase. In the prospective baseline phase, the mean number of weekly cluster headache attacks was 17.5, and was similar across treatment groups.
EMGALITY 300 mg demonstrated statistically significant improvements for efficacy endpoints compared to placebo, as summarized in Table 4 .
| EMGALITY 300 mg N = 49 | Placebo N = 57 | |
| Mean Reduction in Weekly Cluster Headache Attack Frequency (over Weeks 1 to 3) | ||
| Prospective Baseline Cluster Headache Attack Frequency | 17.8 | 17.3 |
| Mean change from baseline | -8.7 | -5.2 |
| Difference from placebo | -3.5 | |
| p-value | 0.036 | |
| ≥50% Weekly Cluster Headache Attack Frequency Responders (at Week 3) | ||
| % Responders | 71.4% | 52.6% |
| Difference from placebo | 18.8% | |
| p-value | 0.046 | |
Figure 7: Mean Change in Weekly Cluster Headache Attack Frequency over Weeks 1 to 3 in Study 4 a

a Abbreviations: BL = baseline; LS = least square; SE = standard error.
Figure 8 shows the distribution of the average percent change from baseline in weekly cluster headache attack frequency across Weeks 1 to 3 in bins of 25%, by treatment group, in Study 4.
Figure 8: Distribution of the Average Percent Change from Baseline in Weekly Cluster Headache Attack Frequency over Weeks 1 to 3 in Study 4 a

a N = number of intent to treat patients with non-missing average percentage change from baseline in weekly cluster headache attack frequency over weeks 1 to 3.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
EMGALITY (galcanezumab-gnlm) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow to slightly brown solution for subcutaneous administration.
EMGALITY is not made with natural rubber latex.
EMGALITY is supplied as follows:
| Pack Size | NDC | |
| Prefilled pen | ||
| 120 mg/mL single-dose | Carton of 1 | 0002-1436-11 |
| 120 mg/mL single-dose | Carton of 2 | 0002-1436-27 |
| Prefilled syringe | ||
| 100 mg/mL single-dose | Carton of 3 | 0002-3115-09 |
| 120 mg/mL single-dose | Carton of 1 | 0002-2377-11 |
| 120 mg/mL single-dose | Carton of 2 | 0002-2377-27 |
Storage and Handling
- Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect EMGALITY from light until use.
- Do not freeze.
- Do not shake.
- EMGALITY may be stored out of refrigeration in the original carton at temperatures up to 30°C (86°F) for up to 7 days. Once stored out of refrigeration, do not place back in the refrigerator.
- If these conditions are exceeded, EMGALITY must be discarded.
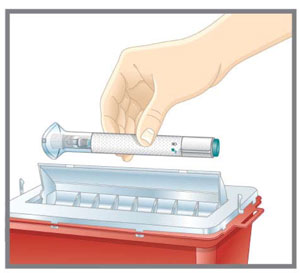
- Discard the EMGALITY single-dose prefilled pen or syringe after use in a puncture-resistant container.
| INSTRUCTIONS FOR USE |
| EMGALITY ® [em-GAL-it-ē] |
| (galcanezumab-gnlm) |
| injection, for subcutaneous use |
| Prefilled Pen |
| This Instructions for Use contains information on how to inject EMGALITY |
 |
| This Instructions for Use is for patients with migraine. |
| For subcutaneous injection only. |
| Important Information You Need to Know Before Injecting EMGALITY |
|
|
|
|
|
|
|
| INSTRUCTIONS FOR USE |
| Before you use the EMGALITY Pen, read and carefully follow all the step-by-step instructions. |
| Parts of the EMGALITY Pen |
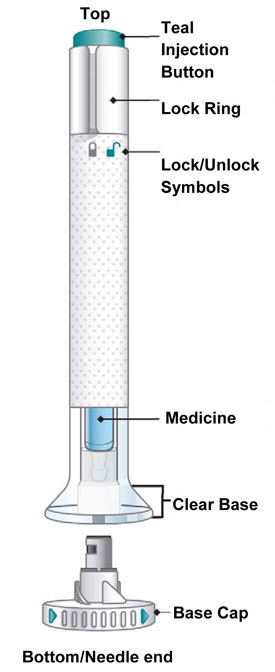 |
| Step 1 Preparing to Inject EMGALITY | |
Step 1a Take the Pen from the refrigerator Check your prescription.
Put the original package with any unused Pens back in the refrigerator. Leave the base cap on until you are ready to inject. Leave the Pen at room temperature for 30 minutes before injecting. Do not microwave the Pen, run hot water over it, or leave it in direct sunlight. Do not shake. | |
Step 1b Gather Supplies For each injection you will need:
| |
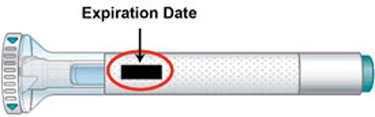
Step 1c Inspect the Pen and the medicine Make sure you have the right medicine. The medicine inside should be clear. Its color may be colorless to slightly yellow to slightly brown. Do not use the Pen, and throw away (dispose of) as directed by your healthcare provider or pharmacist if:
| |
 | |
Step 1d Prepare for injection Wash your hands with soap and water before you inject your EMGALITY. Make sure a sharps disposal container is close by. | |
Step 1e Choose your injection site Your healthcare provider can help you choose the injection site that is best for you. | |
 |
|
| |
| |
| |
| |
| |
| Step 2 Injecting EMGALITY | ||||
| Step 2a Uncap the Pen | 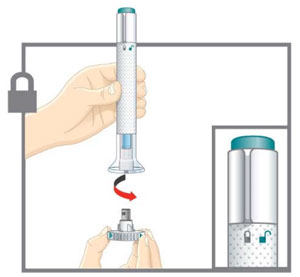 | |||
 | Make sure the Pen is locked. Leave the base cap on until you are ready to inject. | |||
| ||||
| ||||
| ||||
| Step 2b Place and Unlock | 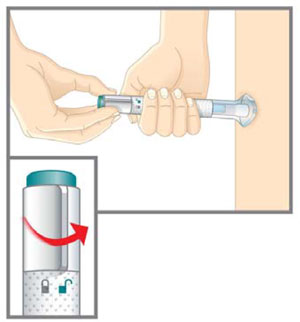 | |||
| ||||
 | Turn the lock ring to the unlock position. | |||
| Step 2c Press and Hold for 10 Seconds | 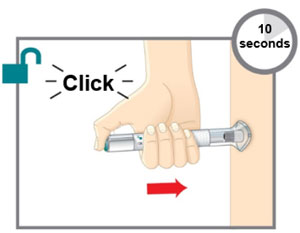 | |||
| ||||
| ||||
|  | |||
Step 3 Disposing of EMGALITY | ||
| Step 3a Throw away the used Pen | ||
|  | |
| ||
| Commonly Asked Questions | ||
| Q. | What if I see bubbles in the Pen? | |
| A. | It is normal to have air bubbles in the Pen. EMGALITY is injected under your skin (subcutaneous injection), so these air bubbles will not harm you. | |
| Q. | What if there is a drop of liquid on the tip of the needle when I remove the base cap? | |
| A. | It is okay to see a drop of liquid on the tip of the needle. | |
| Q. | What if I unlocked the Pen and pressed the teal injection button before I twisted off the base cap? | |
| A. | Do not remove the base cap. Dispose of the Pen and get a new one. | |
| Q. | Do I need to hold the injection button down until the injection is complete? | |
| A. | This is not necessary, but it may help you keep the Pen steady and firm against your skin. | |
| Q. | What if the needle did not retract after my injection? | |
| A. | Do not touch the needle or replace the base cap. Store in a safe place to avoid an accidental needlestick and contact 1-800-545-5979 for instructions on how to return the Pen. | |
| Q. | What if there is a drop of liquid or blood on my skin after my injection? | |
| A. | This is normal. Press a cotton ball or gauze over the injection site. Do not rub the injection site. | |
| Q. | What if I heard more than 2 clicks during my injection – 2 loud clicks and a soft one. Did I get my complete injection? | |
| A. | Some patients may hear a soft click right before the second loud click. That is the normal operation of the Pen. Do not remove the Pen from your skin until you hear the second loud click. | |
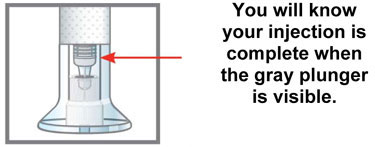
| Q. | How can I tell if my injection is complete? | |
| A. | After you press the teal injection button, you will hear 2 loud clicks. The second click tells you that your injection is complete. You will also see the gray plunger at the top of the clear base. | |
| If you have more questions about how to use the EMGALITY Pen: | ||
|  | |
Storing EMGALITY | |
| |
| |
| |
| |
| |
| |
| |
| Read the full Prescribing Information and Patient Information for EMGALITY inside this box to learn more about your medicine. | |
| Eli Lilly and Company Indianapolis, IN 46285, USA | ||
| US License Number 1891 | ||
| EMGALITY ® is a registered trademark of Eli Lilly and Company. | ||
| Copyright © 2018, 2025, Eli Lilly and Company. All rights reserved. | ||
| The Pen meets the current dose accuracy and functional requirements of ISO 11608-1 and 11608-5. This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: 11/2025 | ||
| EMG-0006-AI-120MG-IFU-20251119 |
Mechanism of Action
Galcanezumab-gnlm is a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP) ligand and blocks its binding to the receptor.