Get your patient on Humatrope (Somatropin)
Humatrope patient education
Patient toolkit
Dosage & administration

Humatrope prescribing information
| Warning and Precautions, Slipped Capital Femoral Epiphysis in Pediatric Patients (5.10 ) | 11/2025 |
INDICATIONS AND USAGE
HUMATROPE is a recombinant human growth hormone indicated for:
- Pediatric Patients: growth failure due to inadequate secretion of endogenous growth hormone (GH); short stature associated with Turner syndrome; Idiopathic Short Stature (ISS), height standard deviation score (SDS) <-2.25, and associated with growth rates unlikely to permit attainment of adult height in the normal range; short stature or growth failure in short stature homeobox-containing gene (SHOX) deficiency; short stature born small for gestational age (SGA) with no catch-up growth by 2 years to 4 years of age. (1.1 )
- Adult Patients: replacement of endogenous GH in adults with GH deficiency. (1.2 )
Pediatric Patients
HUMATROPE is indicated for the treatment of pediatric patients with:
- growth failure due to inadequate secretion of endogenous growth hormone (GH),
- short stature associated with Turner syndrome,
- Idiopathic Short Stature (ISS), height standard deviation score (SDS) <-2.25, and associated with growth rates unlikely to permit attainment of adult height in the normal range,
- short stature or growth failure in short stature homeobox-containing gene (SHOX) deficiency,
- short stature born small for gestational age (SGA) with no catch-up growth by 2 years to 4 years of age.
Adult Patients
HUMATROPE is indicated for the replacement of endogenous GH in adults with GH deficiency.
DOSAGE AND ADMINISTRATION
- Administer by subcutaneous injection to the back of upper arm, abdomen, buttock, or thigh with regular rotation of injection sites. (2.1 )
- Pediatric Dosage - divide the calculated weekly dosage into equal doses given either 6, or 7 days per week.
- Adult Dosage - Either of the following two dosing regimens may be used:
- Non-weight based dosing: Initiate with a dose of approximately 0.2 mg/day (range: 0.15 mg/day-0.3 mg/day) and increase the dose every 1-2 months by increments of approximately 0.1 mg/day-0.2 mg/day, according to individual patient requirements (2.3 )
- Weight-based dosing (Not recommended for obese patients): Initiate at 0.006 mg/kg daily and increase the dose according to individual patient requirements to a maximum of 0.0125 mg/kg daily (2.3 )
- See Full Prescribing Information for reconstitution instructions. (2.4 )
Administration and Use Instructions
- Therapy with HUMATROPE should be supervised by a physician who is experienced in the diagnosis and management of patients with the conditions for which HUMATROPE is indicated [see Indications and Usage (1 )] .
- Fundoscopic examination should be performed routinely before initiating treatment with HUMATROPE to exclude preexisting papilledema, and periodically thereafter [see Warnings and Precautions (5.5 )] .
- Leave HUMATROPE at room temperature for 10 minutes prior to administration.
- Administer HUMATROPE by subcutaneous injection to the back of the upper arm, abdomen, buttock, or thigh with regular rotation of injection sites to avoid lipoatrophy.
Pediatric Dosage
- Individualize dosage for each patient based on the growth response.
- Divide the calculated weekly HUMATROPE dosage into equal doses given either 6 or 7 days per week.
- The recommended weekly dose in milligrams (mg) per kilogram (kg) of body weight for pediatric patients is:
- Pediatric GH Deficiency:0.18 mg/kg/week to 0.3 mg/kg/week (0.026 to 0.043 mg/kg/day)
- Turner Syndrome:Up to 0.375 mg/kg/week (up to.054 mg/kg/day)
- Idiopathic Short Stature:Up to 0.37 mg/kg/week (up to 0.053 mg/kg/day)
- SHOX Deficiency:0.35 mg/kg/week (0.05 mg/kg/day)
- Small for Gestational Age (SGA):Up to 0.47 mg/kg/week (up to 0.067 mg/kg/day)
- In very short pediatric patients, height SDS less than -3, and older pubertal pediatric patients consider initiating treatment with a larger dose of HUMATROPE (up to 0.067 mg/kg/day). Consider a gradual reduction in dosage if substantial catch-up growth is observed during the first few years of therapy. In pediatric patients less than 4 years of age with less severe short stature, baseline height SDS values between -2 and -3, consider initiating treatment at 0.033 mg/kg/day and titrate the dose as needed.
- Assess compliance and evaluate other causes of poor growth such as hypothyroidism, under-nutrition, advanced bone age and antibodies to recombinant human GH if patients experience failure to increase height velocity, particularly during the first year of treatment.
- Discontinue HUMATROPE for stimulation of linear growth once epiphyseal fusion has occurred [see Contraindications (4 )] .
Adult Dosage
- Patients who were treated with somatropin for GH deficiency in childhood and whose epiphyses are closed should be reevaluated before continuation of somatropin for GH deficient adults.
- Consider using a lower starting dose and smaller dose increment increases for geriatric patients as they may be at increased risk for adverse reactions with HUMATROPE than younger individuals [see Use in Specific Populations (8.5 )] .
- Women may require higher doses and patients receiving oral estrogen may require higher doses [see Drug Interactions (7 )] .
- Administer the prescribed dose daily.
- Either of two HUMATROPE dosing regimens may be used:
- Non-weight based:
- Initiate HUMATROPE with a dose of approximately 0.2 mg/day (range, 0.15 mg/day to 0.3 mg/day) and increase the dose every 1-2 months by increments of approximately 0.1 mg/day to 0.2 mg/day, according to individual patient requirements based on the clinical response and serum insulin-like growth factor 1 (IGF-1) concentrations.
- Use the patient's clinical response, adverse reactions, and determination of age- and gender-adjusted serum IGF-1 concentrations as guidance in dose titration.
- Maintenance dosages will vary considerably from person to person, and between male and female patients.
- Weight-based:
- Initiate HUMATROPE at 0.006 mg/kg daily and increase the dose according to individual patient requirements to a maximum of 0.0125 mg/kg daily.
- Use the patient's clinical response, adverse reactions, and determination of age- and gender-adjusted serum IGF-1 concentrations as guidance in dose titration.
- Maintenance dosages will vary considerably from person to person, and between male and female patients
- Not recommended for obese patients as they are more likely to experience adverse reactions with this regimen.
- Non-weight based:
Reconstitution of Cartridges
- Each single-patient-use HUMATROPE cartridge is designed for use only with the appropriate corresponding HumatroPen ® supplied separately.
- Reconstitute each cartridge of HUMATROPE using only the diluent syringe that accompanies the cartridge. Do not shake. The reconstituted solution should be clear.
- Inspect visually for particulate matter and discoloration. If the resulting solution is cloudy or contains particulate matter do not use.
- The somatropin concentrations for the reconstituted HUMATROPE cartridges are as follows in Table 1 :
Cartridge Somatropin Concentration 6 mg 2.08 mg/mL 12 mg 4.17 mg/mL 24 mg 8.33 mg/mL
- Reconstituted cartridges of HUMATROPE are stable for up to 28 days when refrigerated at 36° to 46°F (2° to 8°C). Do not leave at room temperature more than 30 minutes per day. Avoid freezing the reconstituted cartridge. Protect HUMATROPE from light during storage.
- For patients with known hypersensitivity to the diluent supplied with the HUMATROPE cartridge [see Warnings and Precautions (5.6 )] , do not use HUMATROPE cartridge.
DOSAGE FORMS AND STRENGTHS
Each single-patient-use HUMATROPE cartridge is designed for use only with the appropriate corresponding HumatroPen ® supplied separately.
HUMATROPE is a white lyophilized powder available as follows:
- For injection: 6 mg in a single-patient-use cartridge (gold)
- For injection: 12 mg in a single-patient-use cartridge (teal)
- For injection: 24 mg in a single-patient-use cartridge (purple)
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Limited available data with somatropin use in pregnant women are insufficient to determine a drug-associated risk of adverse developmental outcomes. Animal reproduction studies have not been conducted with HUMATROPE.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Lactation
Risk Summary
There is no information regarding the presence of somatropin in human milk. Limited published data indicate that exogenous somatropin does not increase normal breastmilk concentrations of growth hormone. No adverse effects related to somatropin in the breastfed infant have been reported. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for HUMATROPE and any potential adverse effects on the breastfed infant from HUMATROPE or from the underlying maternal condition.
Pediatric Use
Safety and effectiveness of HUMATROPE in pediatric patients have been established in growth failure due to inadequate secretion of endogenous growth hormone, short stature associated with Turner syndrome, idiopathic short stature (ISS), short stature or growth failure in SHOX deficiency, and short stature in children born small for gestational age (SGA) with no catch-up growth by 2 years to 4 years of age.
Growth Failure due to Inadequate Secretion of Endogenous Growth Hormone
Safety and effectiveness of HUMATROPE have been established in pediatric patients with growth failure due to growth hormone deficiency based on data from an open-label, uncontrolled, multicenter study with HUMATROPE in 314 pediatric patients conducted for up to 8 years [see Clinical Studies (14.1 )] .
Short Stature Associated with Turner Syndrome
Safety and effectiveness of HUMATROPE have been established in pediatric patients with short stature associated with Turner syndrome based on data from one long-term, randomized, open-label, multicenter, concurrently controlled study; two long-term, open-label multicenter, historically controlled US studies; and one long-term, randomized, US dose-response study with HUMATROPE in 181 pediatric patients [see Clinical Studies (14.2 )] .
Idiopathic Short Stature (ISS)
Safety and effectiveness of HUMATROPE have been established in pediatric patients with ISS based on data from two randomized, multicenter studies, one placebo-controlled study and one dose-response study with HUMATROPE in 310 pediatric patients [see Clinical Studies (14.3 )] .
Short Stature or Growth Failure in SHOX Deficiency
Safety and effectiveness of HUMATROPE have been established in pediatric patients with short stature or growth failure in SHOX deficiency based on data from a randomized, controlled, two-year, three-arm, open-label study with HUMATROPE in 52 pediatric patients [see Clinical Studies (14.4 )] .
Short Stature in Children Born Small for Gestational Age (SGA) with No Catch-up Growth by 2 Years to 4 Years of Age
Safety and effectiveness of HUMATROPE have been established in pediatric patients with short stature born SGA with no catch-up growth based on data from two clinical studies with HUMATROPE in 214 pediatric patients [see Clinical Studies (14.5 )] .
Geriatric Use
The safety and effectiveness of HUMATROPE in patients aged 65 years and over has not been evaluated in clinical studies. Elderly patients may be more sensitive to the action of somatropin, and therefore may be more prone to development of adverse reactions. A lower starting dose and smaller dose increments should be considered for older patients [see Dosage and Administration (2.3 )] .
CONTRAINDICATIONS
HUMATROPE is contraindicated in patients with:
- Acute critical illness after open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure due to the risk of increased mortality with use of pharmacologic doses of somatropin [see Warnings and Precautions (5.1 )] .
- Pediatric patients with Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea, or have severe respiratory impairment due to the risk of sudden death [see Warnings and Precautions (5.2 )] .
- Active malignancy [see Warnings and Precautions (5.3 )] .
- Known hypersensitivity to somatropin or any of the excipients in HUMATROPE. Systemic hypersensitivity reactions have been reported with postmarketing use of somatropins [see Warnings and Precautions (5.6 )] .
- Active proliferative or severe non-proliferative diabetic retinopathy.
- Pediatric patients with closed epiphyses.
WARNINGS AND PRECAUTIONS
- Increased Risk of Neoplasm: Second neoplasms have occurred in childhood cancer survivors. Monitor patients with preexisting tumors for progression or recurrence. (5.3 )
- Glucose Intolerance and Diabetes Mellitus: HUMATROPE may decrease insulin sensitivity, particularly at higher doses. Monitor glucose levels periodically in all patients receiving HUMATROPE, especially in patients with existing diabetes mellitus or at risk for development. (5.4 )
- Intracranial Hypertension (IH): Has been reported usually within 8 weeks of initiation. Perform fundoscopic examinations prior to initiation and periodically thereafter. If papilledema occurs, stop treatment. (5.5 )
- Hypersensitivity: Serious hypersensitivity reactions may occur. In the event of an allergic reaction, seek prompt medical attention. (5.6 )
- Fluid Retention: May occur in adults and may be dose dependent. (5.7 )
- Hypoadrenalism: Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism. (5.8 )
- Hypothyroidism: Monitor thyroid function periodically as hypothyroidism may occur or worsen after initiation of somatropin. (5.9 )
- Slipped Capital Femoral Epiphysis in Pediatric Patients: May occur; evaluate patients with onset of a limp or hip/knee pain. (5.10 )
- Progression of Preexisting Scoliosis in Pediatric Patients: Monitor patients with scoliosis for progression. (5.11 )
- Pancreatitis: Has been reported; consider pancreatitis in patients with abdominal pain, especially pediatric patients. (5.12 )
Acute Critical Illness
Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin [see Contraindications (4 )] . Two placebo-controlled clinical studies in non-GH deficient adult patients (n=522) with these conditions in intensive care units revealed a significant increase in mortality (42% vs. 19%) among somatropin-treated patients (doses 5.3-8.0 mg/day) compared to those receiving placebo. The safety of continuing HUMATROPE treatment in patients receiving replacement doses for approved indications who concurrently develop these illnesses has not been established. HUMATROPE is not indicated for the treatment of non-GH deficient adults.
Sudden Death in Pediatric Patients with Prader-Willi Syndrome
There have been reports of sudden death after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. Patients with Prader-Willi syndrome should be evaluated for signs of upper airway obstruction and sleep apnea before initiation of treatment with somatropin. If, during treatment with somatropin, patients show signs of upper airway obstruction (including onset of, or increased, snoring) and/or new onset sleep apnea, treatment should be interrupted. All patients with Prader-Willi syndrome treated with somatropin should also have effective weight control and be monitored for signs of respiratory infection, which should be diagnosed as early as possible and treated aggressively [see Contraindications (4 )] . HUMATROPE is not indicated for the treatment of pediatric patients who have growth failure due to Prader-Willi syndrome.
Increased Risk of Neoplasms
Active Malignancy
There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy [see Contraindications (4 )]. Any preexisting malignancy should be inactive and its treatment complete prior to instituting therapy with HUMATROPE. Discontinue HUMATROPE if there is evidence of recurrent activity.
Risk of Second Neoplasm in Pediatric Patients
An increased risk of a second neoplasm in pediatric cancer survivors who were treated with radiation to the brain/head and who developed subsequent GH deficiency and were treated with somatropin has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. In adults, it is unknown whether there is any relationship between somatropin replacement therapy and CNS tumor recurrence. Monitor all patients receiving HUMATROPE who have a history of GH deficiency secondary to an intracranial neoplasm for progression or recurrence of the tumor.
New Malignancy During Treatment
Because pediatric patients with certain rare genetic causes of short stature have an increased risk of developing malignancies, thoroughly consider the risks and benefits of starting HUMATROPE in these patients. If HUMATROPE is initiated, carefully monitor patients for development of neoplasms.
Monitor all patients receiving HUMATROPE carefully for increased growth, or potential malignant changes, of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of pre-existing nevi.
Glucose Intolerance and Diabetes Mellitus
Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. New onset type 2 diabetes mellitus has been reported in patients taking somatropin. Previously undiagnosed impaired glucose tolerance and overt diabetes mellitus may be unmasked. Monitor glucose levels periodically in all patients receiving HUMATROPE, especially in those with risk factors for diabetes mellitus, such as obesity, Turner syndrome, or a family history of diabetes mellitus. Patients with preexisting type 1 or type 2 diabetes mellitus or impaired glucose tolerance should be monitored closely. The doses of antidiabetic agents may require adjustment when HUMATROPE is initiated.
Intracranial Hypertension
Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropins. Symptoms usually occurred within the first eight (8) weeks after the initiation of somatropin therapy. In all reported cases, IH-associated signs and symptoms rapidly resolved after cessation of therapy or a reduction of the somatropin dose. Fundoscopic examination should be performed routinely before initiating treatment with HUMATROPE to exclude preexisting papilledema, and periodically thereafter. If papilledema is observed by fundoscopy during somatropin treatment, treatment should be stopped. If somatropin-induced IH is diagnosed, treatment with HUMATROPE can be restarted at a lower dose after IH-associated signs and symptoms have resolved. Patients with Turner syndrome may be at increased risk for the development of IH.
Severe Hypersensitivity
Serious systemic hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with postmarketing use of somatropins. Patients and caregivers should be informed that such reactions are possible and that prompt medical attention should be sought if an allergic reaction occurs. Do not use HUMATROPE in patients with known hypersensitivity to somatropin or any of the excipients in HUMATROPE. Do not use HUMATROPE cartridges in patients with known hypersensitivity to metacresol or glycerin [see Dosage and Administration (2.4 ), Contraindications (4 )] .
Fluid Retention
Fluid retention during somatropin replacement therapy may frequently occur. Clinical manifestations of fluid retention (e.g. edema, arthralgia, myalgia, nerve compression syndromes including carpal tunnel syndrome/paresthesia) are usually transient and dose dependent.
Hypoadrenalism
Patients receiving somatropin therapy who have or are at risk for pituitary hormone deficiency(s) may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. In addition, patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of HUMATROPE treatment. Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism [see Drug Interactions (7 )].
Hypothyroidism
Undiagnosed/untreated hypothyroidism may prevent an optimal response to HUMATROPE, in particular, the growth response in pediatric patients. Patients with Turner syndrome have an inherently increased risk of developing autoimmune thyroid disease and primary hypothyroidism. In patients with GH deficiency, central (secondary) hypothyroidism may first become evident or worsen during somatropin treatment. Therefore, patients treated with somatropin should have periodic thyroid function tests performed, and thyroid hormone replacement therapy should be initiated or appropriately adjusted when indicated.
Slipped Capital Femoral Epiphysis in Pediatric Patients
Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Slipped capital femoral epiphysis may lead to osteonecrosis. Cases of slipped capital femoral epiphysis with or without osteonecrosis have been reported in pediatric patients with short stature receiving somatropin. Evaluate pediatric patients receiving HUMATROPE with the onset of a limp or complaints of hip or knee pain for slipped capital femoral epiphysis and osteonecrosis and manage accordingly.
Progression of Preexisting Scoliosis in Pediatric Patients
Somatropin increases the growth rate, and progression of existing scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for progression of scoliosis.
Pancreatitis
Cases of pancreatitis have been reported in pediatric patients and adults receiving somatropins. The risk may be greater risk in pediatric patients compared with adults. Published literature indicates that girls who have Turner syndrome may be at greater risk than other pediatric patients receiving somatropins. Pancreatitis should be considered in patients who develop abdominal pain.
Lipoatrophy
When somatropins are administered subcutaneously at the same site over a long period of time, tissue atrophy may result. Rotate injection sites when administering HUMATROPE to reduce this risk [see Dosage and Administration (2.1 )] .
Laboratory Tests
Serum levels of inorganic phosphorus, alkaline phosphatase, parathyroid hormone and IGF-I may increase after HUMATROPE therapy.
ADVERSE REACTIONS
The following important adverse reactions are also described elsewhere in the labeling:
- Increased mortality in patients with acute critical illness [see Warnings and Precautions (5.1 )]
- Fatalities in children with Prader-Willi syndrome [see Warnings and Precautions (5.2 )]
- Neoplasms [see Warnings and Precautions (5.3 )]
- Glucose intolerance and diabetes mellitus [see Warnings and Precautions (5.4 )]
- Intracranial hypertension [see Warnings and Precautions (5.5 )]
- Severe hypersensitivity [see Warnings and Precautions (5.6 )]
- Fluid retention [see Warnings and Precautions (5.7 )]
- Hypoadrenalism [see Warnings and Precautions (5.8 )]
- Hypothyroidism [see Warnings and Precautions (5.9 )]
- Slipped capital femoral epiphysis in pediatric patients [see Warnings and Precautions (5.10 )]
- Progression of preexisting scoliosis in pediatric patients [see Warnings and Precautions (5.11 )]
- Pancreatitis [see Warnings and Precautions (5.12 )]
- Lipoatrophy [see Warnings and Precautions (5.13 )]
Clinical Studies Experience
Because clinical studies are conducted under varying conditions, adverse reaction rates observed during the clinical studies performed with one somatropin formulation cannot always be directly compared to the rates observed during the clinical studies performed with a different somatropin formulation, and may not reflect the adverse reaction rates observed in practice.
Pediatric Patients
Growth Failure Due to Inadequate Secretion of Endogenous Growth Hormone
In an uncontrolled open-label study, 314 treatment-naive children aged >2 years who had GH deficiency were treated with HUMATROPE (0.06 mg/kg 3 times per week) for up to 8 years. Adverse reactions of special interest are reported in Table 2 .
a Dose=0.06 mg/kg 3 times per week for up to 8 years. | |
b n=1 | |
| Adverse Reaction | HUMATROPE a (n=314) |
| Hypothyroidism | 25% |
| Allergic reaction | 11% |
| Arthralgia | 6% |
| Bone disorder | 4% |
| Edema | 4% |
| Injection site pain/reaction | 4% |
| Neoplasm/tumor | 2% |
| Cardiovascular disorders | 1% |
| Thyroid disorders | 1% |
| Intracranial hypertension | 0% b |
Short Stature Associated with Turner Syndrome
In a randomized, concurrent-controlled (untreated), open-label study until attainment of adult height, the adverse reactions of special interest occurring in 74 patients treated with Humatrope at dose 0.3 mg/kg/week (mean duration 4.1 years) and in 62 untreated patients (mean duration 3.7 years) are reported in Table 3 . A similar increase in otitis media was observed in an 18-month placebo-controlled study.
| Untreated (n=62) | HUMATROPE (n=74) | |
| Surgical procedure | 27% | 45% |
| Otitis media | 26% | 43% |
| Ear disorders | 5% | 18% |
Idiopathic Short Stature
Adverse reactions occurring in a randomized, placebo-controlled study of HUMATROPE treatment (0.22 mg/kg/week) until attainment of adult height (mean duration of HUMATROPE treatment 3.7 years, mean duration of placebo treatment 3.3 years) are reported in Table 4 . Mean fasting serum insulin concentration increased by 10% in the HUMATROPE treatment group at the end of treatment relative to baseline, but remained within the normal reference range.
| Placebo (n=31) | HUMATROPE (n=37) | |
| Scoliosis | 13% | 19% |
| Otitis media | 7% | 16% |
| Hyperlipidemia | 3% | 8% |
| Gynecomastia | 3% | 5% |
| Hip pain | 0 | 3% |
| Arthralgia | 3% | 11% |
| Arthrosis | 7% | 11% |
| Myalgia | 13% | 24% |
| Hypertension | 0 | 3% |
In a dose-response study (239 patients treated for 2 years), among HUMATROPE dose groups [0.24 mg/kg/week (n=78), 0.37 mg/kg/week (n=83), 0.24 mg/kg/week for the first year and 0.37 mg/kg/week thereafter (n=78)], mean fasting blood glucose, mean glycosylated hemoglobin, and the incidence of elevated fasting blood glucose concentrations were similar. One patient developed glucose intolerance and high serum HbA 1c .
Short Stature or Growth Failure in SHOX Deficiency
Adverse reactions of special interest from a 2-year randomized, open-label study with HUMATROPE (0.35 mg/kg/wk) compared to no treatment are presented in Table 5 . During the 2-year study period, the proportion of patients who had at least one IGF-I concentration greater than 2.0 SD above the age- and gender-appropriate mean was 10 of 27 (37%) for the HUMATROPE-treated group vs. 0 of 24 patients (0%) for the untreated group. The proportion of patients who had at least one IGFBP-3 concentration greater than 2.0 SD above the age and gender appropriate mean was 16 of 27 (59%) for the HUMATROPE treated group vs. 7 of 24 (29%) for the untreated group.
a Percentage calculated for males only: Untreated (0/1), HUMATROPE (1/12) | ||
| Untreated (n=25) | HUMATROPE (n=27) | |
| Arthralgia | 8% | 11% |
| Gynecomastia a | 0% | 8% |
| Excessive number of cutaneous nevi | 0% | 7% |
| Scoliosis | 0% | 47% |
Small for Gestational Age (SGA) with No Catch-up Growth by Age 2-4 Years
In a 2-year, multicenter, randomized study, 193 pediatric patients were treated with 2 different HUMATROPE treatment regimens: a fixed dose of 0.067 mg/kg/day (FHD group) or an individually adjusted dose regimen (IAD group; starting dose 0.035 mg/kg/day which could be increased as early as Month 3 to 0.067 mg/kg/day based on a validated growth prediction model). Reported adverse reactions included: common childhood infectious diseases, otitis media, headaches, and slipped capital femoral epiphysis (n=1. Six patients (4 in the FHD group and 2 in the IAD group whose dose was increased from 0.035 mg/kg/day to 0.067 mg/kg/day [one at Month 3 and one at Year 1]) had impaired fasting glucose at Year 2. Two of 6 had impaired fasting glucose during the study, and one discontinued HUMATROPE at month 15 as a consequence. At study completion, 20-25% of patients had serum IGF-I SDS values > +2.
The following adverse reactions were reported from an observational study of 340 pediatric patients who received HUMATROPE with an average dosage of 0.041 mg/kg/day (maximum dose: 0.084 mg/kg/day) for an average of 3.0 years: type 2 diabetes mellitus (n=1), carpal tunnel syndrome (n=1) and an exacerbation of preexisting scoliosis (n=1).
Adult Patients with GH deficiency
Adult-Onset GH Deficiency
In the first 6 months of controlled blinded studies during which patients received either HUMATROPE or placebo, patients who received HUMATROPE experienced an increase in edema (17% vs. 4%) and peripheral edema (12% vs. 0%). Edema, muscle pain, joint pain, and joint disorder were reported early in therapy and tended to be transient or responsive to dosage titration.
Two of 113 patients developed carpal tunnel syndrome after beginning maintenance therapy without a low dose (0.00625 mg/kg/day) lead-in phase. Symptoms abated in these patients after dosage reduction.
Adverse reactions with ≥5% overall occurrence rate during 12 or 18 months of replacement therapy with HUMATROPE are shown in Table 6 (adult-onset patients) and in Table 7 (childhood-onset patients).
| Adverse Reaction | 18 Months Exposure [Placebo (6 Months)/Humatrope (12 Months)] (n=44) | 18 Months Humatrope Exposure (n=52) |
| Edema | 11% | 21% |
| Arthralgia | 14% | 17% |
| Paresthesia | 14% | 17% |
| Myalgia | 9% | 14% |
| Pain | 14% | 14% |
| Peripheral edema | 18% | 12% |
| Headache | 7% | 8% |
| Hypertension | 5% | 8% |
| Joint disorder | 2% | 6% |
| Rhinitis | 11% | 13% |
| Back pain | 9% | 10% |
| Acne | 0% | 6% |
| Surgical procedure | 2% | 6% |
| Flu syndrome | 7% | 4% |
Childhood-Onset GH Deficiency
Two double-blind, placebo-controlled studies were conducted in 67 adult patients who had received previous somatropin treatment during childhood. Patients were randomized to receive either placebo injections or HUMATROPE (0.00625 mg/kg/day for the first 4 weeks, then 0.0125 mg/kg/day thereafter) for the first 6 months, followed by open-label use of HUMATROPE for the next 12 months for all patients. During the placebo-controlled phase (first 6 months) of the study, elevations of serum glutamic oxaloacetic transferase were reported more for HUMATROPE-treated (13% vs. 0%) than placebo-treated patients.
a Abbreviations: ALT=alanine aminotransferase; AST=aspartate aminotransferase | ||
| Adverse Reaction | 18 Months Exposure [Placebo (6 Months)/Humatrope (12 Months)] (n=30) | 18 Months Humatrope Exposure (n=32) |
| AST a increased | 7% | 13% |
| Headache | 7% | 9% |
| Asthenia | 3% | 6% |
| Edema | 10% | 6% |
| Myalgia | 7% | 6% |
| Pain | 10% | 6% |
| ALT a increased | 7% | 6% |
| Flu syndrome | 10% | 16% |
| Cough increased | 0% | 6% |
| Hypesthesia | 0% | 6% |
| Rhinitis | 7% | 6% |
| Respiratory disorder | 7% | 3% |
| Gastritis | 7% | 0% |
| Pharyngitis | 7% | 3% |
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of somatropin or HUMATROPE. Because these adverse events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Severe Hypersensitivity Reactions — Serious systemic hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with postmarketing use of somatropins
Neurologic — Headaches (common in pediatric patients and occasional in adults)
Skin — Increase in size or number of cutaneous nevi
Endocrine — Gynecomastia
Gastrointestinal — Pancreatitis
Metabolic — New-onset type 2 diabetes mellitus
Musculoskeletal and connective tissue disorders — Osteonecrosis in pediatric patients
Neoplasia — Leukemia has been reported in a small number of GH deficient pediatric patients treated with somatropin
DRUG INTERACTIONS
Table 8 includes a list of drugs with clinically important drug interactions when administered concomitantly with HUMATROPE and instructions for preventing or managing them.
| Replacement Glucocorticoid Treatment | |
| Clinical Impact: | Microsomal enzyme 11β-hydroxysteroid dehydrogenase type 1 (11βHSD-1) is required for conversion of cortisone to its active metabolite, cortisol, in hepatic and adipose tissue. HUMATROPE inhibits 11βHSD-1. Consequently, individuals with untreated GH deficiency have relative increases in 11βHSD-1 and serum cortisol. Initiation of HUMATROPE may result in inhibition of 11βHSD-1 and reduced serum cortisol concentrations. |
| Intervention: | Patients treated with glucocorticoid replacement for hypoadrenalism may require an increase in their maintenance or stress doses following initiation of HUMATROPE [see Warnings and Precautions (5.8 )]. |
| Examples: | Cortisone acetate and prednisone may be effected more than others since conversion of these drugs to their biologically active metabolites is dependent on the activity of 11βHSD-1. |
| Non-Replacement Glucocorticoid Treatment in Pediatric Patients | |
| Clinical Impact: | Non-replacement glucocorticoid treatment, including supraphysiologic glucocorticoid treatment, may attenuate the growth promoting effects of HUMATROPE in pediatric patients. |
| Intervention: | Carefully adjust glucocorticoid dosing in pediatric patients receiving glucocorticoid treatments to avoid both hypoadrenalism and an inhibitory effect on growth. |
| Cytochrome P450-Metabolized Drugs | |
| Clinical Impact: | Limited published data indicate that somatropin treatment increases cytochrome P450 (CP450)-mediated antipyrine clearance. HUMATROPE may alter the clearance of compounds known to be metabolized by CP450 liver enzymes. |
| Intervention: | Careful monitoring is advisable when HUMATROPE is administered in combination with drugs metabolized by CP450 liver enzymes. |
| Oral Estrogen | |
| Clinical Impact: | Oral estrogens may reduce the serum IGF-1 response to HUMATROPE. |
| Intervention: | Patients receiving oral estrogen may require greater HUMATROPE dosages [see Dosage and Administration (2.3 )] . |
| Insulin and/or Other Hypoglycemic Agents | |
| Clinical Impact: | Treatment with HUMATROPE may decrease insulin sensitivity, particularly at higher doses. |
| Intervention: | Patients with diabetes mellitus may require adjustment of their doses of insulin and/or other hypoglycemic agents [see Warnings and Precautions (5.4 )]. |
DESCRIPTION
Somatropin is a human growth hormone (GH) produced by recombinant DNA technology using Escherichia coli. The protein is comprised of 191 amino acid residues and has a molecular weight of about 22,125 daltons. The amino acid sequence is identical to that of human GH of pituitary origin.
HUMATROPE (somatropin) for injection is a sterile, white, lyophilized powder intended for subcutaneous injection after reconstitution supplied in a cartridge. Phosphoric acid and/or sodium hydroxide may have been added to adjust the pH. Reconstituted solutions have a pH of approximately 7.5. This product is oxygen sensitive.
Cartridge — Each single-patient-use cartridge of HUMATROPE contains either 6 mg (18 IU), 12 mg (36 IU), or 24 mg (72 IU) of somatropin. Each HUMATROPE cartridge contains the following components (see Table 9 ):
| Component | Cartridge | ||
| 6 mg (gold) | 12 mg (teal) | 24 mg (purple) | |
| Somatropin | 6 mg | 12 mg | 24 mg |
| Dibasic sodium phosphate | 1.36 mg | 2.72 mg | 5.43 mg |
| Glycine | 6 mg | 12 mg | 24 mg |
| Mannitol | 18 mg | 36 mg | 72 mg |
Each cartridge is co-packaged with an accompanying syringe containing approximately 3 mL of diluent containing Water for Injection with 0.3% metacresol as a preservative and 1.7%, 0.29%, and 0.29% glycerin in the 6, 12, and 24 mg cartridges, respectively.
CLINICAL PHARMACOLOGY
Mechanism of Action
Somatropin binds to dimeric GH receptors located within the cell membranes of target tissue cells. This interaction results in intracellular signal transduction and subsequent induction of transcription and translation of GH-dependent proteins including IGF-1, IGF BP-3 and acid-labile subunit. Somatropin has direct tissue and metabolic effects or mediated indirectly by IGF-1, including stimulation of chondrocyte differentiation, and proliferation, stimulation of hepatic glucose output, protein synthesis and lipolysis.
Somatropin stimulates skeletal growth in pediatric patients with GHD as a result of effects on the growth plates (epiphyses) of long bones. The stimulation of skeletal growth increases linear growth rate (height velocity) in most somatropin-treated pediatric patients. Linear growth is facilitated in part by increased cellular protein synthesis.
Pharmacodynamics
Subcutaneous administration of a single dose of Humatrope (0.033 mg/kg body weight) in healthy volunteers (10 males, 10 females) resulted in an increased median IGF-1 level from 202 ng/mL (men) and 107 ng/mL (women) predose to maximal level of 362 ng/mL (men) and 234 ng/mL (women) after a median of 21 hours (men) and 14 hours (women).
Pharmacokinetics
Absorption
HUMATROPE has been studied following intramuscular, subcutaneous, and intravenous administration in healthy adult subjects. A single subcutaneous dose administration of HUMATROPE 0.1 mg/kg (0.27 IU/kg) in healthy subjects (n= 8) resulted in a mean (SD) C max of 63.3 (18.2) ng/mL at median T max of 4.0 (range 3 to 8) hours. The absolute bioavailability of somatropin is 75% after subcutaneous administration.
Distribution
The mean (SD) volume of distribution of somatropin after single dose subcutaneous injection of 0.1 mg/kg (0.27 IU/kg) in healthy subjects is about 0.96 (0.3) L/kg.
Elimination
Metabolism — Extensive metabolism studies have not been conducted. The metabolic fate of somatropin involves classical protein catabolism in both the liver and kidneys. In renal cells, at least a portion of the breakdown products of somatropin is returned to the systemic circulation.
Excretion — In healthy subjects, mean somatropin clearance is 0.18 (0.03) L/hr/kg following subcutaneous administration. The mean half-life of subcutaneous somatropin is 3.8 (1.4) hours. The long half-life observed after subcutaneous administration is due to slow absorption from the injection site. Urinary excretion of intact HUMATROPE has not been measured. Small amounts of somatropin have been detected in the urine of pediatric patients following replacement therapy.
Specific Populations
Geriatric patients — The pharmacokinetics of HUMATROPE have not been studied in patients greater than 65 years of age.
Pediatric patients — The pharmacokinetics of HUMATROPE in pediatric patients are similar to those of adults.
Male and Female Patients — No gender-specific pharmacokinetic studies have been performed with HUMATROPE. The available literature indicates that the pharmacokinetics of somatropin are similar in men and women.
Patients with Renal or hepatic impairment — No studies have been performed with HUMATROPE.
Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of HUMATROPE or other somatropins.
In a clinical study with HUMATROPE during the first 6 months of HUMATROPE therapy in 314 naive patients, 1.6% developed specific antibodies to HUMATROPE (binding capacity ≥0.02 mg/L). None had antibody concentrations which exceeded 2 mg/L. Throughout 8 years of this same study, two patients (0.6%) had binding capacity >2 mg/L. Neither patient demonstrated a decrease in growth velocity at or near the time of increased antibody production. It has been reported that growth attenuation from pituitary-derived GH may occur when antibody concentrations are >1.5 mg/L.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity, mutagenicity and impairment of fertility studies have not been conducted with HUMATROPE.
CLINICAL STUDIES
Pediatric Patients with Growth Failure Due to Inadequate Secretion of Endogenous Growth Hormone
An open-label, uncontrolled, multicenter study with Humatrope was conducted in, 314 treatment-naive children aged >2 years who had GH deficiency. Patients were treated with HUMATROPE (0.06 mg/kg 3 times per week) for up to 8 years. The efficacy of HUMTROPE was assessed by evaluating height velocity. Mean increase in height velocity from baseline of 3.6 ± 1.9 cm/year to 8.8 ± 2.3 cm/year was achieved by 1 year of treatment. Height velocity remained increased above baseline for a small subset of patients (n=12) who remained on-study up to 8 years during the long-term extension phase (6.3 ± 2.5 cm/year at year 8 of treatment).
Pediatric Patients with Short Stature Associated with Turner Syndrome
One long-term, randomized, open-label, Canadian multicenter, concurrently controlled study, two long-term, open-label multicenter, historically controlled US studies and one long-term, randomized, US dose-response study were conducted to evaluate the efficacy of HUMATROPE for treatment of short stature due to Turner syndrome.
The randomized, open-label, Canadian study (NCT00191113) compared near-adult height outcomes for HUMATROPE-treated patients to those of a concurrent control group who received no injections. The HUMATROPE-treated patients received a dosage of 0.3 mg/kg/week given in divided doses 6 times per week from a mean age of 11.7 years for a mean duration of 4.7 years. Puberty was induced with a standardized estrogen regimen initiated at 13 years of age for both treatment groups.
In two of the US studies the effect of long-term treatment with Humatrope (0.375 mg/kg/week given in divided doses either 3 times per week or daily) on adult height was determined by comparing adult heights in the treated patients with those of age-matched historical controls with Turner syndrome who received no growth-promoting therapy. Puberty was induced with a standardized estrogen regimen initiated after 14 years of age in one study; in the second study patients treated early (before 11 years of age) were randomized to begin pubertal induction at either age 12 (n=26) or 15 (n=29) years (conjugated estrogens, 0.3 mg escalating to 0.625 mg daily); those whose treatment was initiated after 11 years of age began estrogen replacement after 1 year of treatment with HUMATROPE.
In the third US study, a randomized, blinded dose-response study, patients were treated from a mean age of 10.9 years for a mean duration of 5.5 years with a weekly HUMATROPE dosage of either 0.27 mg/kg or 0.36 mg/kg administered in divided doses 3 or 6 times weekly.
In summary, patients with Turner syndrome (total n=249 from the 4 studies above) treated to adult height achieved average height gains ranging from 5.0 to 8.3 cm. See Table 10 .
a Data shown are mean values. | |||||||
b RCT: randomized controlled trial; MHT: matched historical controlled trial; RDT: randomized dose-response trial. | |||||||
c Analysis of covariance (ANCOVA) vs. controls: ANCOVA models with GH (0.27 or 0.36 mg/kg/wk) included covariates of baseline age (all studies), baseline height (all studies), study site (all studies), mid-parental height (all studies) and karyotype (US1 and US2 only), baseline bone age (US2 only) or estrogen (low dose or placebo for US3 only). | |||||||
d Group A: GH age <11 yr, estrogen is initiated at age 15. | |||||||
e Group B: GH age <11 yr, estrogen is initiated at age 12. | |||||||
f Group C: GH age >11 yr, estrogen is initiated at after 12 month of treatment with HUMATROPE. | |||||||
| Study | Group | Study Design b | Somatropin Treated Number at Adult Height | GH Age (yr) | Estrogen Age (yr) | GH Duration (yr) | Adult Height Gain (cm) c |
| Canadian | RCT | 27 | 11.7 | 13 | 4.7 | 5.4 | |
| US 1 | MHT | 17 | 9.1 | 15.2 | 7.6 | 7.4 | |
| US 2 | A d | MHT | 29 | 9.4 | 15 | 6.1 | 8.3 |
| B e | MHT | 26 | 9.6 | 12.3 | 5.6 | 5.9 | |
| C f | MHT | 51 | 12.7 | 13.7 | 3.8 | 5 | |
| US 3 | RDT | 99 | 10.9 | 8-13.5 | 5.5 | 5.7 | |
Pediatric Patients with Idiopathic Short Stature (ISS)
Two randomized, multicenter studies, one placebo-controlled and one dose-response, were conducted in pediatric patients with idiopathic short stature. The diagnosis of idiopathic short stature was made after excluding other known causes of short stature, as well as GH deficiency. The placebo-controlled study enrolled 71 pediatric patients (55 males, 16 females) 9 to 15 years old (mean age 12.4 ± 1.5 years), with short stature, 68 of whom received placebo (n=31) or HUMATROPE (n=37); NCT00191074. Patients were predominately prepubertal (Tanner I, 45%) or in early puberty (Tanner II, 47%) at baseline. In this double-blind study, patients received subcutaneous injections of either HUMATROPE 0.222 mg/kg/week, or placebo given in divided doses 3 times per week until height velocity decreased to ≤1.5 cm/year (“final height”). Final height measurements were available for 33 subjects (22 treated with HUMATROPE, 11 treated with placebo) after a mean treatment duration of 4.4 years (range 0.1-9.1 years).
Results are presented in Table 11 . The number of patients whose final height was above the 5th percentile of the general population height standard for age and sex was significantly greater in the HUMATROPE group than the placebo group (41% vs. 0%), as was the number of patients who gained at least 1 SDS unit in height across the duration of the study (50% vs. 0%).
a For final height population. | ||||
b Abbreviations: BPH=baseline predicted height; CI=confidence interval; FH=final height; NA=not applicable; SDS=standard deviation score. | ||||
c Assessed using a one-way analysis of variance (ANOVA) with effect for treatment. | ||||
d Placebo n=10 as one patient was missing BPH. | ||||
e Assessed using analysis of covariance (ANCOVA) with baseline predicted height SDS as the covariate. | ||||
| Placebo (n=11) Mean (SD) | HUMATROPE (n=22) Mean (SD) | Treatment Effect Mean (95%CI b ) | p-value | |
| Baseline height SDS | -2.8 (0.6) | -2.7 (0.6) | NA b | 0.77 c |
| BPH SDS | -2.3 (0.8) | -2.1 (0.7) | NA | 0.53 c,d |
| Final height SDS | -2.3 (0.6) | -1.8 (0.8) | 0.51 (0.10, 0.92) | 0.017 e |
| FH b SDS - baseline height SDS | 0.4 (0.2) | 0.9 (0.7) | 0.51 (0.04, 0.97) | 0.034 e |
| FH SDS - BPH SDS | -0.1 (0.6) | 0.3 (0.6) | 0.46 (0.02, 0.89) | 0.043 d,e |
The dose-response study included 239 pediatric patients (158 males, 81 females), 5 to 15 years old, (mean age 9.8 ± 2.3 years). Mean ± SD baseline characteristics included: height SDS -3.21 ± 0.70, predicted adult height SDS -2.63 ± 1.08, and height velocity SDS -1.09 ± 1.15. All but 3 patients were prepubertal. Patients were randomized to one of three HUMATROPE treatment groups: 0.24 mg/kg/week (n=78); 0.24 mg/kg/week for 1 year, followed by 0.37 mg/kg/week (n=78); and 0.37 mg/kg/week (n=83). Height velocity was assessed in each treatment group during the first 2 years of therapy. After completing the initial 2-year dose-response phase of the study, 50 patients were followed to final height.
After 2 years of treatment, patients who received the HUMATROPE dosage of 0.37 mg/kg/week (n=71) had a significantly greater increase in mean height velocity than patients who received 0.24 mg/kg/week (n=68), (4.04 vs. 3.27 cm/year respectively (ANOVA p=0.003). A total of 12 patients who received 0.37 mg/kg/week and 10 patients who received 0.24 mg/kg/week treatment had missing 2 year treatment data. The mean difference between final height and baseline predicted height was 7.2 cm for patients who received Humatrope 0.37 mg/kg/week and 5.4 cm for patients who received 0.24 mg/kg/week. The results are presented in Table 12 . While no patient had height above the 5th percentile in any dosage group at baseline, 82% (n=14) of the 17 patients who received 0.37 mg/kg/week and 47% (n=8) of the 17 patients who received 0.24 mg/kg/week achieved final heights above the 5th percentile of the general population height standards.
a Abbreviations: FH=final height; PH=predicted height; CI=confidence interval; cm=centimeters. | |||||
| Placebo-controlled Study 3x per week dosing | Dose Response Study 6x per week dosing | ||||
| Placebo (n=10) | HUMATROPE 0.22 mg/kg (n=22) | HUMATROPE 0.24 mg/kg (n=13) | HUMATROPE 0.24/0.37 mg/kg (n=13) | HUMATROPE 0.37 mg/kg (n=13) | |
| FH - Baseline PH Mean (95% CI), cm | -0.7 (-3.6, 2.3) | +2.2 (0.4, 3.9) | +5.4 (2.8, 7.9) | +6.7 (4.1, 9.2) | +7.2 (4.6, 9.8) |
Pediatric Patients with Short Stature or Growth Failure in SHOX Deficiency
A randomized, controlled, two-year, three-arm, open-label study was conducted to evaluate the efficacy of HUMATROPE for treatment of short stature in pediatric patients with SHOX deficiency who were not GH–deficient; NCT00190658. A total of 52 patients (24 male, 28 female) with SHOX deficiency, 3 to 12.3 years of age, were randomized to either a HUMATROPE-treated arm (27 patients; mean age 7.3 ± 2.1 years) or an untreated control arm (25 patients; mean age 7.5 ± 2.7 years). To determine the comparability of treatment effect between patients with SHOX deficiency and patients with Turner syndrome, the third study arm enrolled 26 patients with Turner syndrome, 4.5 to 11.8 years of age (mean age 7.5 ± 1.9 years) who received HUMATROPE treatment. All patients were prepubertal at study entry. Patients in the HUMATROPE-treated group(s) received daily subcutaneous injections of 0.05 mg/kg of HUMATROPE, equivalent to 0.35 mg/kg/week. Patients in the untreated group received no injections. One untreated patient discontinued the study during the first year. The results are presented in Table 13 ; the mean first-year height velocity of treated patients with SHOX deficiency was significantly greater than that of the untreated patients (mean between-group difference = 3.5 [95% CI: 2.8 – 4.2] cm/year, p<0.001).
a Positive values favor HUMATROPE | ||||
b p<0.001 (assessed using analysis of covariance [ANCOVA] with covariates of treatment Group, Léri-Weill syndrome, sex and baseline age). | ||||
c p<0.001 (assessed using ANCOVA with covariates of treatment group and baseline height [cm]). | ||||
d p<0.001 (assessed using ANCOVA with covariates of treatment group and baseline height SDS). | ||||
e p=0.003 calculated using Fisher's exact test. | ||||
| SHOX Deficiency | Turner Syndrome | |||
| Untreated (n=24) Mean (SD) | HUMATROPE (n=27) Mean (SD) | Treatment Difference a Mean (95% CI) | HUMATROPE (n=26) Mean (SD) | |
| Height Velocity (cm/yr) | ||||
| 1 st Year | 5.2 (1.1) | 8.7 (1.6) b | +3.5 (2.8, 4.2) | 8.9 (2.0) |
| 2 nd Year | 5.4 (1.2) | 7.3 (1.1) b | +2.0 (1.3, 2.6) | 7.0 (1.1) |
| Height Gain (cm) | ||||
| Baseline to 1 st Year | 5.4 (1.2) | 9.1 (1.5) c | +3.7 (2.9, 4.5) | 8.9 (1.9) |
| Baseline to 2 nd Year | 10.5 (1.9) | 16.4 (2.0) c | +5.8 (4.6, 7.1) | 15.7 (2.7) |
| Height SDS Gain | ||||
| Baseline to 1 st Year | 0.1 (0.5) | 0.7 (0.5) d | +0.5 (0.3, 0.8) | 0.8 (0.5) |
| Baseline to 2 nd Year | 0.2 (0.5) | 1.2 (0.7) d | +1.0 (0.7, 1.3) | 1.2 (0.7) |
| Patients with height SDS > -2.0 at 2 years | 1 (4%) | 11 (41%) e | -- | 8 (31%) |
Pediatric Patients with Short Stature Born Small for Gestational Age (SGA) Who Fail to Demonstrate Catch-up Growth by Age 2 - 4 Years
The height increases would be considered similar if the lower bound of the 95% confidence interval (CI) for the mean difference between the groups (IAD – FHD) was greater than -0.5 height SDS. A 2-year, open-label, multicenter, European study enrolled 193 prepubertal, non-GH deficient children with mean chronological age 6.8 ± 2.4 years (range: 3 to 12.3); NCT00191529. Study entry criteria included birth weight <10th percentile and/or birth length SDS <-2 for gestational age, and height SDS for chronological age ≤-3. Exclusion criteria included syndromal conditions (e.g., Turner syndrome), chronic disease (e.g., diabetes mellitus), and active tumors. The primary objective was to compare the increase from baseline in height SDS after 1 year of treatment when HUMATROPE is administered according to an individually adjusted dose (IAD) regimen with a fixed high dose (FHD) regimen. Patients were randomized to either a FHD (0.067 mg/kg/day [0.47 mg/kg/week]; n=99) or an IAD treatment group (n=94). The initial HUMATROPE dosage in the IAD treatment group was 0.035 mg/kg/day (0.25 mg/kg/week). The dosage was increased to 0.067 mg/kg/day in those patients in the IAD group whose 1-year height gain predicted at Month 3 was <0.75 height SDS (n=40) or whose actual height gain measured at Year 1 was <0.75 height SDS (n=11).
The results are presented in Table 14 , The increase from baseline in height SDS in the IAD group was non-inferior to that in the FHD group at Year 1 (mean between-group difference = -0.3 SDS [95% CI: -0.4, -0.2 SDS]). The results were similar when children who entered puberty during the study were removed from the analysis. Data is shown for the efficacy analysis population that included all patients who received at least 1 dose of HUMATROPE and had at least 1 post randomization height measurement. Approximately 85% of the randomized patients completed 2 years of therapy.
a Abbreviations: IAD=individually adjusted dose; FHD=fixed high dose; SD=standard deviation; SDS=standard deviation score | |||
b Least squares mean difference ± standard error and 95% confidence interval based on ANCOVA model with treatment and gender as fixed effects, and baseline height SDS, baseline chronological age, baseline bone age, and mid-parental target height SDS as covariates. | |||
c Only children with actual height measurements were included in the Year 1 and Year 2 analyses. | |||
| IAD Group 0.035 to 0.067 mg/kg/day Mean (SD) | FHD Group 0.067 mg/kg/day Mean (SD) | IAD – FHD Group Difference ± SE (95% CI) b | |
| Baseline | (n=86) -3.9 (0.6) | (n=93) -3.9 (0.7) | -0.0 ± 0.1 (-0.2, 0.2) |
| Year 1 Height SDS Change from baseline | (n=86) c -3.0 (0.7) 0.9 (0.4) | (n=93) c -2.7 (0.7) 1.1 (0.4) | -0.3 ± 0.1 (-0.4, -0.2) p-value <0.001 |
| Year 2 Height SDS Change from baseline | (n=82) c -2.5 (0.8) 1.4 (0.5) | (n=88) c -2.2 (0.7) 1.6 (0.5) | -0.3 ± 0.1 (-0.4, -0.1) |
An open-label, multicenter, single arm study was conducted in France, during which 35 prepubertal, non-GH deficient children were treated for 2 years with HUMATROPE 0.067 mg/kg/day (0.47 mg/kg/week). Mean chronological age at baseline was 9.3 ± 0.9 years (range: 6.7 to 10.8). Additional study entry criteria included birth length SDS <-2 or <3rd percentile for gestational age, and height SDS for chronological age <-2. Exclusion criteria included syndromal conditions (e.g., Turner syndrome), and chronic disease (e.g., diabetes mellitus). All 35 patients completed the study. Mean height SDS increased from a baseline value of -2.7 (SD 0.5) to -1.5 (SD 0.6) after 2 years of HUMATROPE treatment.
Adult Patients with Growth Hormone Deficiency
Two multicenter studies in patients with adult-onset GH deficiency (n=98) and two studies in patients with childhood-onset GH deficiency (n=67) were designed to assess the effects of replacement therapy with HUMATROPE. Adult-onset patients and childhood-onset patients differed by diagnosis (organic vs. idiopathic pituitary disease), body size (average vs. small [mean height (171 cm vs 161 cm) and weight (85 kg vs 64 kg)]), and age (mean 44 vs. 29 years).Each study included a 6-month randomized, blinded, placebo-controlled phase, during which approximately half of the patients received placebo injections, while the other half received HUMATROPE injections. The HUMATROPE dosages for all studies were identical: 1 month of treatment at 0.00625 mg/kg/day followed by 0.0125 mg/kg/day for the next 5 months. The 6-month, double-blind phase was followed by 12 months of open-label HUMATROPE treatment for all patients. The primary efficacy measures were body composition (lean body mass and fat mass) and lipid parameters [Total cholesterol, high-density lipoprotein (HDL) and low-density lipoprotein (LDL)]. Lean body mass was determined by bioelectrical impedance analysis (BIA), validated with potassium 40. Body fat was assessed by BIA and sum of skinfold thickness. Lipid subfractions were analyzed by standard assay methods in a central laboratory.
In a study with patients with adult-onset GH deficiency, HUMATROPE treatment (vs. placebo) resulted in an increase in mean lean body mass (2.59 vs. -0.22 kg, p<0.001) and a decrease in body fat (-3.27 vs. 0.56 kg, p<0.001). Similar changes were seen in childhood-onset GH deficient patients. These changes in lean body mass persisted throughout the 18-month period for both the adult-onset and childhood-onset groups; the changes in fat mass persisted in the childhood-onset group. Serum concentrations of HDL cholesterol which were low at baseline (mean, 31.0 mg/dL and 33.9 mg/dL in adult-onset (n=46) and childhood-onset (n=30) patients, respectively) had increased by the end of 18 months of HUMATROPE treatment (mean change of 13.7 and 11.1 mg/dL for the adult-onset (n=40) and childhood-onset (n=21) groups, respectively p<0.001; there was no adjustment for missing data). Total cholesterol and LDL did not show significant difference from baseline results by the end of 18 months of HUMATROPE treatment.
In an additional 2-year, open-label, randomized study 149 patients with childhood-onset GH deficiency who had completed pediatric somatropin therapy, had attained final height (height velocity <1 cm/yr) and were confirmed to be GH-deficient as young adults, were randomized to receive HUMATROPE 0.0125 mg/kg/day (n=59), HUMATROPE 0.025 mg/kg/day (n=58), or no treatment (control) (n=32). Total bone mineral content (BMC) increased by 5.2% ± 3.9% in the control group (n=28 for those with BMC measurements), 7.0% ± 7.2% in the HUMATROPE 0.025 mg/kg/day group (n=51) and 8.0% ± 8.9% in the HUMATROPE 0.0125 mg/kg/day group (n=51). For the treatment effect versus control group, p=0.012 (ANOVA); there was no statistically significant difference between the 2 HUMATROPE dose groups. A significant overall treatment effect (ANOVA, p=0.037) was seen for the percentage change in lumbar spine BMC, but not for femoral neck or hip.
HOW SUPPLIED/STORAGE AND HANDLING
Each single-patient-use HUMATROPE cartridge is designed for use only with the appropriate corresponding HumatroPen ® supplied separately.
How Supplied
HUMATROPE (somatropin) for injection is a white lyophilized powder available in the following cartridge sizes in Table 15 :
| NDC | Kit | HUMATROPE | Diluent |
| NDC 0002-8147-01 | Cartridge Kit | 6 mg Single Patient-Use cartridge (gold) | prefilled syringe of Diluent for HUMATROPE |
| NDC 0002-8148-01 | Cartridge Kit | 12 mg Single Patient-Use cartridge (teal) | prefilled syringe of Diluent for HUMATROPE |
| NDC 0002-8149-01 | Cartridge Kit | 24 mg Single Patient-Use cartridge (purple) | prefilled syringe of Diluent for HUMATROPE |
Storage and Handling
Cartridges
Refrigerate cartridges of HUMATROPE and Diluent for HUMATROPE at 36° to 46°F (2° to 8°C). Avoid freezing Diluent for HUMATROPE. Store in the original carton to protect HUMATROPE from light.
Humatrope Cartrige Kit Instuctions for Use
Instructions for Use
HUMATROPE ® (HU-ma-trope)
(somatropin)
For injection, for subcutaneous use
Single-Patient-Use Cartridge Kit
HUMATROPE cartridges are only to be used with the appropriate corresponding HumatroPen ® injection devices supplied separately.
Read this Instructions for Use before you start using HUMATROPE Cartridge Kit and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your or your child's medical condition or treatment.
Your healthcare provider should show you how to prepare, mix, measure, and inject HUMATROPE the right way before you use it for the first time. Ask your healthcare provider if you have any questions.
How should I store HUMATROPE?
- Store HUMATROPE in the refrigerator between 36°F to 46°F (2°C to 8°C) before and after mixing.
- Do not freeze.
- Store HUMATROPE in the original carton to protect from light.
- HUMATROPE that has been mixed and is in liquid form must be used within 28 days. Throw away any mixed HUMATROPE left over after 28 days.
- Before giving an injection, check the expiration date on the Cartridge. Do not use the Cartridge if it has expired (See Figure A ).
Important:
- HUMATROPE Cartridges should not be used if you are allergic to metacresol or glycerin.
- The HUMATROPE Cartridge is not recommended for use by blind or visually impaired individuals without the help of a sighted individual trained on how to use it.
- If your healthcare provider changes the prescribed cartridge strength you will need to get a new HumatroPen to match the new cartridge strength.
- Only use the following HUMATROPE cartridges with the following HumatroPen:
- 6 mg cartridge with HumatroPen 6 mg
- 12 mg cartridge with HumatroPen 12 mg
- 24 mg cartridge with HumatroPen 24 mg
HUMATROPE Cartridge Kit (See Figure A )
- 1 Cartridge with 6 mg, 12 mg, or 24 mg of powdered HUMATROPE
- 1 prefilled syringe with Diluent (the liquid used to mix the powdered HUMATROPE)
Note: There are 3 strengths of HUMATROPE Cartridges that have different amounts of HUMATROPE (6 mg, 12 mg, or 24 mg). Make sure that you have the Cartridge that your healthcare provider prescribed.
Mixing the HUMATROPE in the Cartridge
Only use the prefilled Diluent Syringe that comes with your HUMATROPE Cartridge Kit to mix the HUMATROPE in the Cartridge. Do not use the diluent that comes in the HUMATROPE vial box, or any other liquid.
HUMATROPE Cartridge Kit mixing Instructions
Parts (Figure A) |
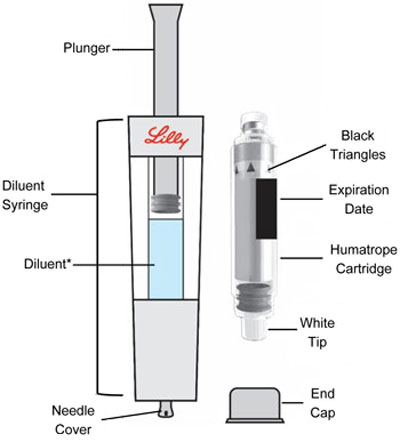 |
Only use a HUMATROPE Cartridge Kit to prepare the HUMATROPE Cartridge.
• Note : The liquid (Diluent) should be clear, colorless, and free of particles and is shown as blue in the figure (See Figure A ).
| Preparing Your New HUMATROPE Cartridge | |||
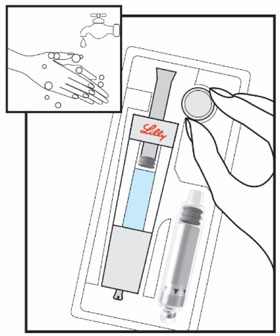 | 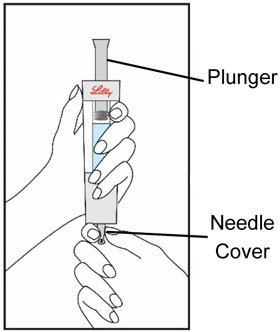 | 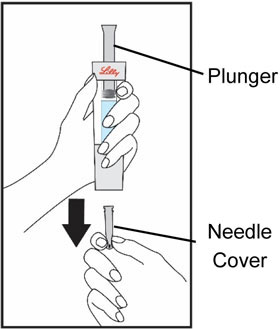 | |
| Start by thoroughly washing your hands. Remove all contents from the tray. Note: The HUMATROPE Cartridge is made for left or right handed use so you may handle it the way that is most comfortable for you. | Hold the gray needle cover at the bottom of the Diluent Syringe. | Pull the needle cover straight down to remove from the Diluent Syringe and throw it away. Do not push the plunger yet. It is okay if a drop of Diluent is seen. You do not have to release air from the Diluent Syringe. | |
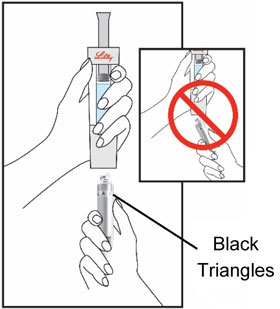 | 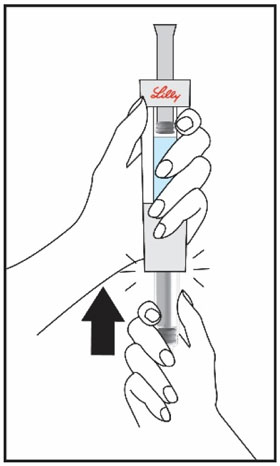 |  | |
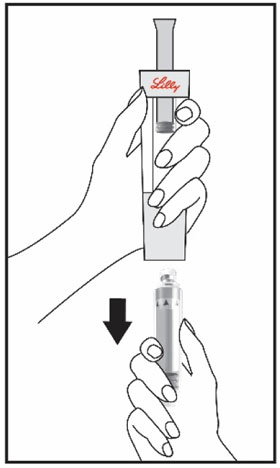
| Hold the Cartridge, with the black triangles up toward the Diluent Syringe. Make sure the Cartridge and Diluent Syringe are facing each other in a straight line. Do not insert the Cartridge at an angle. | Push the Cartridge straight into the Diluent Syringe until it stops and the black triangles are covered inside the Diluent Syringe. You may hear or feel a click. Do not twist the Cartridge. | Hold the Diluent Syringe and the Cartridge together with both hands . Push and release the plunger 2 or 3 times until the Diluent is in the Cartridge. | |
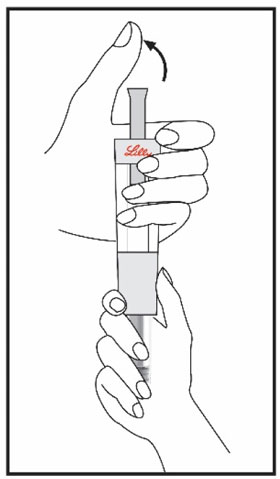 |  | 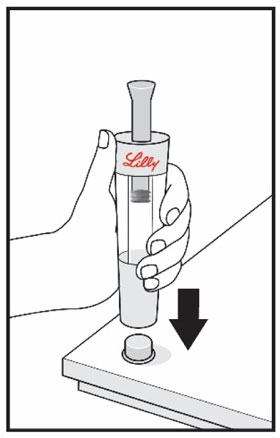 | |
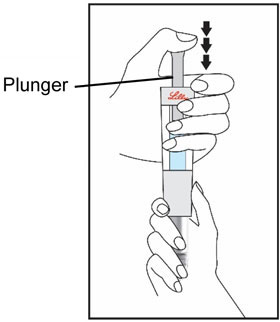
| Remove your thumb from the plunger and check that the Diluent Syringe is empty. It is normal for small drops of Diluent to remain in the Diluent Syringe. | With your thumb off the plunger, pull the Cartridge away from the Diluent Syringe. | Place the end cap on a hard, flat surface. Push the Diluent Syringe onto the end cap and throw away the Diluent Syringe right away as instructed by your healthcare provider. | |
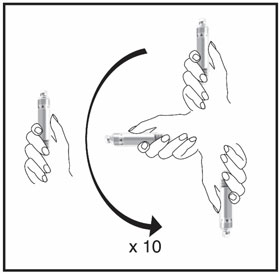 |  | ||
| Mix the medicine in the Cartridge by gently moving it in an up and down motion 10 times. Then let the Cartridge sit for 3 minutes. Do not shake. | Check the HUMATROPE liquid, it should be clear. If the liquid is clear, your Cartridge is now prepared and ready to be attached to your HumatroPen (See the Instructions for Use for HumatroPen 6 mg, 12 mg or 24 mg). If the liquid is cloudy or contains particles, gently mix the medicine in the Cartridge 10 more times. Let the Cartridge sit for 5 more minutes. If the liquid still remains cloudy or contains particles, do not use the Cartridge. Contact your healthcare provider or call Lilly at 1-800-545-5979 or go to www.humatrope.com. If you have questions about preparing your HUMATROPE Cartridge, you should contact your healthcare provider. | ||
Talk to your healthcare provider about the right way to use your HUMATROPE Cartridge Kit and HumatroPen injection device, the right sites to inject HUMATROPE, and how to rotate your injection sites.
Disposing of used Diluent Syringes:
- Put your used Diluent Syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. Do not reuse or share your needles or syringes with other people. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal .
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
HUMATROPE ® and HumatroPen ® are registered trademarks of Eli Lilly and Company.
Copyright © 2019, 2025 Eli Lilly and Company. All rights reserved.
Revised: November 2025
Manufactured by: Eli Lilly and Company, Indianapolis, IN 46285, USA
US License Number 1891
www.HUMATROPE.com
HTRCART-0003-IFU-20251125
Mechanism of Action
Somatropin binds to dimeric GH receptors located within the cell membranes of target tissue cells. This interaction results in intracellular signal transduction and subsequent induction of transcription and translation of GH-dependent proteins including IGF-1, IGF BP-3 and acid-labile subunit. Somatropin has direct tissue and metabolic effects or mediated indirectly by IGF-1, including stimulation of chondrocyte differentiation, and proliferation, stimulation of hepatic glucose output, protein synthesis and lipolysis.
Somatropin stimulates skeletal growth in pediatric patients with GHD as a result of effects on the growth plates (epiphyses) of long bones. The stimulation of skeletal growth increases linear growth rate (height velocity) in most somatropin-treated pediatric patients. Linear growth is facilitated in part by increased cellular protein synthesis.