Get your patient on Onfi (Clobazam)
Onfi patient education
Patient toolkit
Dosage & administration

Onfi prescribing information
WARNING: RISKS FROM CONCOMITANT USE WITH OPIOIDS; ABUSE, MISUSE, AND ADDICTION; and DEPENDENCE AND WITHDRAWAL REACTIONS
- Concomitant use of benzodiazepines and opioids may result in profound sedation, respiratory depression, coma, and death. Reserve concomitant prescribing of these drugs for patients for whom alternative treatment options are inadequate. Limit dosages and durations to the minimum required. Follow patients for signs and symptoms of respiratory depression and sedation [see Warnings and Precautions (5.1 ), Drug Interactions (7.1 )] .
- The use of benzodiazepines, including ONFI, exposes users to risks of abuse, misuse, and addiction, which can lead to overdose or death. Abuse and misuse of benzodiazepines commonly involve concomitant use of other medications, alcohol, and/or illicit substances, which is associated with an increased frequency of serious adverse outcomes. Before prescribing ONFI and throughout treatment, assess each patient’s risk for abuse, misuse, and addiction [see Warnings and Precautions (5.2 )] .
- The continued use of benzodiazepines, including ONFI, may lead to clinically significant physical dependence. The risks of dependence and withdrawal increase with longer treatment duration and higher daily dose. Abrupt discontinuation or rapid dosage reduction of ONFI after continued use may precipitate acute withdrawal reactions, which can be life-threatening. To reduce the risk of withdrawal reactions, use a gradual taper to discontinue ONFI or reduce the dosage [see Dosage and Administration (2.2 ) and Warnings and Precautions (5.3 )] .
1 INDICATIONS AND USAGE
ONFI ® (clobazam) is indicated for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome (LGS) in patients 2 years of age or older.
2 DOSAGE AND ADMINISTRATION
Dosing Information
A daily dose of ONFI greater than 5 mg should be administered in divided doses twice daily; a 5 mg daily dose can be administered as a single dose. Dose patients according to body weight. Individualize dosing within each body weight group, based on clinical efficacy and tolerability. Each dose in Table 1 (e.g., 5 to 20 mg in ≤30 kg weight group) has been shown to be effective, although effectiveness increases with increasing dose [see Clinical Studies (14 )] . Do not proceed with dose escalation more rapidly than weekly, because serum concentrations of clobazam and its active metabolite require 5 and 9 days, respectively, to reach steady-state.
| ≤30 kg Body Weight | >30 kg Body Weight | |
| Starting Dose | 5 mg | 10 mg |
| Starting Day 7 | 10 mg | 20 mg |
| Starting Day 14 | 20 mg | 40 mg |
Discontinuation or Dosage Reduction of ONFI
To reduce the risk of withdrawal reactions, increased seizure frequency, and status epilepticus, use a gradual taper to discontinue ONFI or reduce the dosage. Taper by decreasing the total daily dose by 5-10 mg/day on a weekly basis until discontinued. If a patient develops withdrawal reactions, consider pausing the taper or increasing the dosage to the previous tapered dosage level. Subsequently decrease the dosage more slowly [see Warnings and Precautions (5.3 ) and Drug Abuse and Dependence (9.3 )] .
Important Administration Instructions
Instruct patients to read the "Instructions for Use" carefully for complete directions on how to properly dose and administer ONFI oral suspension.
ONFI Tablet Oral Administration ONFI tablets can be taken with or without food. ONFI tablets can be administered whole, broken in half along the score, or crushed and mixed in applesauce.
ONFI Oral Suspension Oral Administration ONFI oral suspension can be taken with or without food [see Clinical Pharmacology (12.3 )] .
Shake ONFI Oral Suspension well before every administration. When administering the oral suspension, use only the oral dosing syringe provided with the product. Each carton includes two syringes, but only one syringe should be used for dosing. The second oral syringe is reserved as a replacement in case the first syringe is damaged or lost. Insert the provided adapter firmly into the neck of the bottle before first use and keep the adapter in place for the duration of the usage of the bottle. To withdraw the dose, insert the dosing syringe into the adapter and invert the bottle then slowly pull back the plunger to prescribed dose. After removing the syringe from the bottle adapter, slowly squirt ONFI Oral Suspension into the corner of the patient's mouth. Replace the cap after each use. The cap fits over the adapter when the adapter is properly placed. See ONFI Oral Suspension "Instructions for Use" for complete instruction on how to properly dose and administer the ONFI Oral Suspension.
Dosage Adjustments in Geriatric Patients
Plasma concentrations at any given dose are generally higher in the elderly: proceed slowly with dose escalation. The starting dose should be 5 mg/day for all elderly patients. Then titrate elderly patients according to weight, but to half the dose presented in Table 1, as tolerated. If necessary and based upon clinical response, an additional titration to the maximum dose (20 mg/day or 40 mg/day, depending on weight) may be started on day 21 [see Use in Specific Populations (8.5 )] .
Dosage Adjustments in CYP2C19 Poor Metabolizers
In CYP2C19 poor metabolizers, levels of N-desmethylclobazam, clobazam's active metabolite, will be increased. Therefore, in patients known to be CYP2C19 poor metabolizers, the starting dose should be 5 mg/day and dose titration should proceed slowly according to weight, but to half the dose presented in Table 1, as tolerated. If necessary and based upon clinical response, an additional titration to the maximum dose (20 mg/day or 40 mg/day, depending on the weight group) may be started on day 21 [see Use in Specific Populations (8.6 ), Clinical Pharmacology (12.5 )] .
Patients with Renal Impairment
No dose adjustment is required for patients with mild and moderate renal impairment. There is no experience with ONFI in patients with severe renal impairment or end stage renal disease (ESRD). It is not known if clobazam or its active metabolite, N-desmethylclobazam, is dialyzable [see Use in Specific Populations (8.7 ), Clinical Pharmacology (12.3 )] .
Dosage Adjustments in Patients with Hepatic Impairment
ONFI is hepatically metabolized; however, there are limited data to characterize the effect of hepatic impairment on the pharmacokinetics of ONFI. For this reason, proceed slowly with dosing escalations. For patients with mild to moderate hepatic impairment (Child-Pugh score 5-9), the starting dose should be 5 mg/day in both weight groups. Then titrate patients according to weight, but to half the dose presented in Table 1, as tolerated. If necessary and based upon clinical response, start an additional titration on day 21 to the maximum dose (20 mg/day or 40 mg/day, depending on the weight group). There is inadequate information about metabolism of ONFI in patients with severe hepatic impairment. Therefore no dosing recommendation in those patients can be given [see Use in Specific Populations (8.8 ), Clinical Pharmacology (12.3 )] .
3 DOSAGE FORMS AND STRENGTHS
Tablets: 10 mg and 20 mg with a functional score for oral administration. Each ONFI tablet is a white to off-white, oval tablet with a functional score on one side and either a "1" and "0" or a "2" and "0" debossed on the other side.
Oral Suspension: 2.5 mg/mL for oral administration. Each bottle contains 120 mL of an off-white suspension.
8 USE IN SPECIFIC POPULATIONS
Pregnancy: Based on animal data, may cause fetal harm (8.1 )
Pregnancy
Pregnancy Registry There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to AEDs, such as ONFI, during pregnancy. Healthcare providers are encouraged to recommend that pregnant women taking ONFI enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry by calling 1-888-233-2334 or online at http://www.aedpregnancyregistry.org/ .
Risk Summary Neonates born to mothers using benzodiazepines late in pregnancy have been reported to experience symptoms of sedation and/or neonatal withdrawal [see Warnings and Precautions (5.9) and Clinical Considerations] . Available data from published observational studies of pregnant women exposed to benzodiazepines do not report a clear association with benzodiazepines and major birth defects ( see Data ).
Administration of clobazam to pregnant rats and rabbits during the period of organogenesis or to rats throughout pregnancy and lactation resulted in developmental toxicity, including increased incidences of fetal malformations and mortality, at plasma exposures for clobazam and its major active metabolite, N-desmethylclobazam, below those expected at therapeutic doses in patients [ see Animal Data ]. Data for other benzodiazepines suggest the possibility of long-term effects on neurobehavioral and immunological function in animals following prenatal exposure to benzodiazepines at clinically relevant doses. ONFI should be used during pregnancy only if the potential benefit to the mother justifies the potential risk to the fetus. Advise a pregnant woman and women of childbearing age of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations Fetal/Neonatal Adverse Reactions Benzodiazepines cross the placenta and may produce respiratory depression, hypotonia, and sedation in neonates. Monitor neonates exposed to ONFI during pregnancy or labor for signs of sedation, respiratory depression, hypotonia, and feeding problems. Monitor neonates exposed to ONFI during pregnancy for signs of withdrawal. Manage these neonates accordingly [see Warnings and Precautions (5.9 )] .
Data Human Data Published data from observational studies on the use of benzodiazepines during pregnancy do not report a clear association with benzodiazepines and major birth defects. Although early studies reported an increased risk of congenital malformations with diazepam and chlordiazepoxide, there was no consistent pattern noted. In addition, the majority of more recent case-control and cohort studies of benzodiazepine use during pregnancy, which were adjusted for confounding exposures to alcohol, tobacco and other medications, have not confirmed these findings.
Animal Data In a study in which clobazam (0, 150, 450, or 750 mg/kg/day) was orally administered to pregnant rats throughout the period of organogenesis, embryofetal mortality and incidences of fetal skeletal variations were increased at all doses. The low-effect dose for embryofetal developmental toxicity in rats (150 mg/kg/day) was associated with plasma exposures (AUC) for clobazam and its major active metabolite, N-desmethylclobazam, lower than those in humans at the maximum recommended human dose (MRHD) of 40 mg/day.
Oral administration of clobazam (0, 10, 30, or 75 mg/kg/day) to pregnant rabbits throughout the period of organogenesis resulted in decreased fetal body weights, and increased incidences of fetal malformations (visceral and skeletal) at the mid and high doses, and an increase in embryofetal mortality at the high dose. Incidences of fetal variations were increased at all doses. The highest dose tested was associated with maternal toxicity (ataxia and decreased activity). The low-effect dose for embryofetal developmental toxicity in rabbits (10 mg/kg/day) was associated with plasma exposures for clobazam and N-desmethylclobazam lower than those in humans at the MRHD.
Oral administration of clobazam (0, 50, 350, or 750 mg/kg/day) to rats throughout pregnancy and lactation resulted in increased embryofetal mortality at the high dose, decreased pup survival at the mid and high doses and alterations in offspring behavior (locomotor activity) at all doses. The low-effect dose for adverse effects on pre- and postnatal development in rats (50 mg/kg/day) was associated with plasma exposures for clobazam and N-desmethylclobazam lower than those in humans at the MRHD.
8.2 Lactation
Risk Summary ONFI is excreted in human milk ( see Data) . There are reports of sedation, poor feeding and poor weight gain in infants exposed to benzodiazepines through breast milk. There are no data on the effects of clobazam on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ONFI and any potential adverse effects on the breastfed infant from ONFI or from the underlying maternal condition.
Clinical Considerations Adverse reactions such as somnolence and difficulty feeding have been reported in infants during breastfeeding in postmarketing experience with ONFI. Infants exposed to ONFI through breast milk should be monitored for sedation, poor feeding and poor weight gain.
Data Scientific literature on ONFI use during lactation is limited. After short-term administration, clobazam and N-desmethylclobazam are transferred into breast milk.
Females and Males of Reproductive Potential
Administration of clobazam to rats prior to and during mating and early gestation resulted in adverse effects on fertility and early embryonic development at plasma exposures for clobazam and its major active metabolite, N-desmethylclobazam, below those in humans at the MRHD [see Nonclinical Toxicology (13.1 )] .
Pediatric Use
Safety and effectiveness in patients less than 2 years of age have not been established.
In a study in which clobazam (0, 4, 36, or 120 mg/kg/day) was orally administered to rats during the juvenile period of development (postnatal days 14 to 48), adverse effects on growth (decreased bone density and bone length) and behavior (altered motor activity and auditory startle response; learning deficit) were observed at the high dose. The effect on bone density, but not on behavior, was reversible when drug was discontinued. The no-effect level for juvenile toxicity (36 mg/kg/day) was associated with plasma exposures (AUC) to clobazam and its major active metabolite, N-desmethylclobazam, less than those expected at therapeutic doses in pediatric patients.
Geriatric Use
Clinical studies of ONFI did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. However, elderly subjects appear to eliminate clobazam more slowly than younger subjects based on population pharmacokinetic analysis. For these reasons, the initial dose in elderly patients should be 5 mg/day. Patients should be titrated initially to 10-20 mg/day. Patients may be titrated further to a maximum daily dose of 40 mg if tolerated [see Dosage and Administration (2.4 ), Clinical Pharmacology (12.3 )] .
CYP2C19 Poor Metabolizers
Renal Impairment
The pharmacokinetics of ONFI were evaluated in patients with mild and moderate renal impairment. There were no significant differences in systemic exposure (AUC and C max ) between patients with mild or moderate renal impairment and healthy subjects. No dose adjustment is required for patients with mild and moderate renal impairment. There is essentially no experience with ONFI in patients with severe renal impairment or ESRD. It is not known if clobazam or its active metabolite, N-desmethylclobazam, is dialyzable [see Dosage and Administration (2.6 ), Clinical Pharmacology (12.3 )] .
Hepatic Impairment
ONFI is hepatically metabolized; however, there are limited data to characterize the effect of hepatic impairment on the pharmacokinetics of ONFI. For this reason, dosage adjustment is recommended in patients with mild to moderate hepatic impairment (Child-Pugh score 5-9). There is inadequate information about metabolism of ONFI in patients with severe hepatic impairment [see Dosage and Administration (2.7 ), Clinical Pharmacology (12.3 )] .
5 WARNINGS AND PRECAUTIONS
Risks from Concomitant Use with Opioids
Concomitant use of benzodiazepines, including ONFI, and opioids may result in profound sedation, respiratory depression, coma, and death. Because of these risks, reserve concomitant prescribing of benzodiazepines and opioids for patients for whom alternative treatment options are inadequate.
Observational studies have demonstrated that concomitant use of opioid analgesics and benzodiazepines increases the risk of drug-related mortality compared to use of opioids alone. If a decision is made to prescribe ONFI concomitantly with opioids, prescribe the lowest effective dosages and minimum durations of concomitant use, and follow patients closely for signs and symptoms of respiratory depression and sedation. Advise both patients and caregivers about the risks of respiratory depression and sedation when ONFI is used with opioids [see Drug Interactions (7.1 )] .
Abuse, Misuse, and Addiction
The use of benzodiazepines, including ONFI, exposes users to the risks of abuse, misuse, and addiction, which can lead to overdose or death. Abuse and misuse of benzodiazepines often (but not always) involve the use of doses greater than the maximum recommended dosage and commonly involve concomitant use of other medications, alcohol, and/or illicit substances, which is associated with an increased frequency of serious adverse outcomes, including respiratory depression, overdose, or death [see Drug Abuse and Dependence (9.2 )] .
Before prescribing ONFI and throughout treatment, assess each patient’s risk for abuse, misuse, and addiction (e.g., using a standardized screening tool). Use of ONFI, particularly in patients at elevated risk, necessitates counseling about the risks and proper use of ONFI along with monitoring for signs and symptoms of abuse, misuse, and addiction. Prescribe the lowest effective dosage; avoid or minimize concomitant use of CNS depressants and other substances associated with abuse, misuse, and addiction (e.g., opioid analgesics, stimulants); and advise patients on the proper disposal of unused drug. If a substance use disorder is suspected, evaluate the patient and institute (or refer them for) early treatment, as appropriate.
Dependence and Withdrawal Reactions
To reduce the risk of withdrawal reactions, use a gradual taper to discontinue ONFI or reduce the dosage [see Dosage and Administration (2.2 )] .
Patients at an increased risk of withdrawal adverse reactions after benzodiazepine discontinuation or rapid dosage reduction include those who take higher dosages, and those who have had longer durations of use.
Acute Withdrawal Reactions
The continued use of benzodiazepines, including ONFI, may lead to clinically significant physical dependence. Abrupt discontinuation or rapid dosage reduction of ONFI after continued use, or administration of flumazenil (a benzodiazepine antagonist) may precipitate acute withdrawal reactions, which can be life-threatening (e.g., seizures) [see Drug Abuse and Dependence (9.3 )] .
Protracted Withdrawal Syndrome
In some cases, benzodiazepine users have developed a protracted withdrawal syndrome with withdrawal symptoms lasting weeks to more than 12 months [see Drug Abuse and Dependence (9.3 )] .
Potentiation of Sedation from Concomitant Use with Central Nervous System Depressants
Since ONFI has a central nervous system (CNS) depressant effect, patients or their caregivers should be cautioned against simultaneous use with other CNS depressant drugs or alcohol, and cautioned that the effects of other CNS depressant drugs or alcohol may be potentiated [see Drug Interactions (7.2 )] .
Somnolence or Sedation
ONFI causes somnolence and sedation. In clinical trials, somnolence or sedation was reported at all effective doses and was dose-related.
In general, somnolence and sedation begin within the first month of treatment and may diminish with continued treatment. Prescribers should monitor patients for somnolence and sedation, particularly with concomitant use of other central nervous system depressants. Prescribers should caution patients against engaging in hazardous activities requiring mental alertness, such as operating dangerous machinery or motor vehicles, until the effect of ONFI is known.
Serious Dermatological Reactions
Serious skin reactions, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), have been reported with ONFI in both children and adults during the postmarketing period. Patients should be closely monitored for signs or symptoms of SJS/TEN, especially during the first 8 weeks of treatment initiation or when re-introducing therapy. ONFI should be discontinued at the first sign of rash, unless the rash is clearly not drug-related. If signs or symptoms suggest SJS/TEN, use of this drug should not be resumed and alternative therapy should be considered [see Contraindications (4 )] .
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as multiorgan hypersensitivity, has been reported in patients taking antiepileptic drugs, including ONFI. These events can be fatal or life-threatening, particularly if diagnosis and treatment do not occur as early as possible. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy, and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis, sometimes resembling an acute viral infection. Eosinophilia is often present. Because this disorder is variable in its expression, other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. ONFI should be discontinued if an alternative etiology for the signs or symptoms cannot be established [see Contraindications (4) ] .
Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including ONFI, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted relative risk 1.8, 95% confidence interval [CI]: 1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed. Table 2 shows absolute and relative risk by indication for all evaluated AEDs.
| Indication | Placebo Patients with Events per 1000 Patients | Drug Patients with Events per 1000 Patients | Relative Risk: Incidence of Drug Events in Drug Patients/Incidence in Placebo Patients | Risk Difference: Additional Drug Patients with Events per 1000 Patients |
| Epilepsy | 1.0 | 3.4 | 3.5 | 2.4 |
| Psychiatric | 5.7 | 8.5 | 1.5 | 2.9 |
| Other | 1.0 | 1.8 | 1.9 | 0.9 |
| Total | 2.4 | 4.3 | 1.8 | 1.9 |
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing ONFI or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
Neonatal Sedation and Withdrawal Syndrome
Use of ONFI late in pregnancy can result in sedation (respiratory depression, lethargy, hypotonia) and/or withdrawal symptoms (hyperreflexia, irritability, restlessness, tremors, inconsolable crying, and feeding difficulties) in the neonate [see Use in Specific Populations (8.1) ] . Monitor neonates exposed to ONFI during pregnancy or labor for signs of sedation and monitor neonates exposed to ONFI during pregnancy for signs of withdrawal; manage these neonates accordingly.
6 ADVERSE REACTIONS
Clinically significant adverse reactions that appear in other sections of the labeling include the following:
- Risks from Concomitant Use with Opioids [see Warnings and Precautions (5.1 )]
- Abuse, Misuse, and Addiction [see Warnings and Precautions (5.2 )]
- Dependence and Withdrawal Reactions [see Warnings and Precautions (5.3 )]
- Potentiation of Sedation from Concomitant Use with Central Nervous System Depressants [see Warnings and Precautions (5.4 )]
- Somnolence or Sedation [see Warnings and Precautions (5.5 )]
- Serious Dermatological Reactions [see Contraindications (4 ), Warnings and Precautions (5.6 )]
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity [see Warnings and Precautions (5.7 )]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.8) ]
- Neonatal Sedation and Withdrawal Syndrome [see Warnings and Precautions (5.9) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
During its development for the adjunctive treatment of seizures associated with LGS, ONFI was administered to 333 healthy volunteers and 300 patients with a current or prior diagnosis of LGS, including 197 patients treated for 12 months or more. The conditions and duration of exposure varied greatly and included single- and multiple-dose clinical pharmacology studies in healthy volunteers and two double-blind studies in patients with LGS (Study 1 and 2) [see Clinical Studies (14 )] . Only Study 1 included a placebo group, allowing comparison of adverse reaction rates on ONFI at several doses to placebo.
Adverse Reactions Leading to Discontinuation in an LGS Placebo Controlled Clinical Trial (Study 1) The adverse reactions associated with ONFI treatment discontinuation in ≥1% of patients in decreasing order of frequency included lethargy, somnolence, ataxia, aggression, fatigue, and insomnia.
Most Common Adverse Reactions in an LGS Placebo Controlled Clinical Trial (Study 1) Table 3 lists the adverse reactions that occurred in ≥5% of ONFI-treated patients (at any dose), and at a rate greater than placebo-treated patients, in the randomized, double-blind, placebo-controlled, parallel group clinical study of adjunctive AED therapy for 15 weeks (Study 1).
| a Maximum daily dose of 5 mg for ≤30 kg body weight; 10 mg for >30 kg body weight b Maximum daily dose of 10 mg for ≤30 kg body weight; 20 mg for >30 kg body weight c Maximum daily dose of 20 mg for ≤30 kg body weight; 40 mg for >30 kg body weight | |||||
| Placebo N=59 % | ONFI Dose Level | All ONFI N=179 % | |||
| Low a N=58 % | Medium b N=62 % | High c N=59 % | |||
| Gastrointestinal Disorders | |||||
| Vomiting | 5 | 9 | 5 | 7 | 7 |
| Constipation | 0 | 2 | 2 | 10 | 5 |
| Dysphagia | 0 | 0 | 0 | 5 | 2 |
| General Disorders and Administration Site Conditions | |||||
| Pyrexia | 3 | 17 | 10 | 12 | 13 |
| Irritability | 5 | 3 | 11 | 5 | 7 |
| Fatigue | 2 | 5 | 5 | 3 | 5 |
| Infections and Infestations | |||||
| Upper respiratory tract infection | 10 | 10 | 13 | 14 | 12 |
| Pneumonia | 2 | 3 | 3 | 7 | 4 |
| Urinary tract infection | 0 | 2 | 5 | 5 | 4 |
| Bronchitis | 0 | 2 | 0 | 5 | 2 |
| Metabolism and Nutrition Disorders | |||||
| Decreased appetite | 3 | 3 | 0 | 7 | 3 |
| Increased appetite | 0 | 2 | 3 | 5 | 3 |
| Nervous System Disorders | |||||
| Somnolence or Sedation | 15 | 17 | 27 | 32 | 26 |
| Somnolence | 12 | 16 | 24 | 25 | 22 |
| Sedation | 3 | 2 | 3 | 9 | 5 |
| Lethargy | 5 | 10 | 5 | 15 | 10 |
| Drooling | 3 | 0 | 13 | 14 | 9 |
| Ataxia | 3 | 3 | 2 | 10 | 5 |
| Psychomotor hyperactivity | 3 | 3 | 3 | 5 | 4 |
| Dysarthria | 0 | 2 | 2 | 5 | 3 |
| Psychiatric Disorders | |||||
| Aggression | 5 | 3 | 8 | 14 | 8 |
| Insomnia | 2 | 2 | 5 | 7 | 5 |
| Respiratory Disorders | |||||
| Cough | 0 | 3 | 5 | 7 | 5 |
Postmarketing Experience
These reactions are reported voluntarily from a population of uncertain size; therefore, it is not possible to estimate their frequency or establish a causal relationship to drug exposure. Adverse reactions are categorized by system organ class.
Blood Disorders: Anemia, eosinophilia, leukopenia, thrombocytopenia Eye Disorders: Diplopia, vision blurred Gastrointestinal Disorders: Abdominal distention General Disorders and Administration Site Conditions: Hypothermia Investigations: Hepatic enzyme increased Musculoskeletal: Muscle spasms Psychiatric Disorders: Agitation, anxiety, apathy, confusional state, depression, delirium, delusion, hallucination Renal and Urinary Disorders: Urinary retention Respiratory Disorders: Aspiration, respiratory depression Skin and Subcutaneous Tissue Disorders: Rash, urticaria, angioedema, and facial and lip edema
7 DRUG INTERACTIONS
Opioids
The concomitant use of benzodiazepines and opioids increases the risk of respiratory depression because of actions at different receptor sites in the CNS that control respiration. Benzodiazepines interact at GABA A sites, and opioids interact primarily at mu receptors. When benzodiazepines and opioids are combined, the potential for benzodiazepines to significantly worsen opioid-related respiratory depression exists. Limit dosage and duration of concomitant use of benzodiazepines and opioids, and follow patients closely for respiratory depression and sedation [see Warnings and Precautions (5.1 )] .
CNS Depressants and Alcohol
Concomitant use of ONFI with other CNS depressants may increase the risk of sedation and somnolence [see Warnings and Precautions (5.4 )] .
Alcohol, as a CNS depressant, will interact with ONFI in a similar way and also increases clobazam's maximum plasma exposure by approximately 50%. Therefore, caution patients or their caregivers against simultaneous use with other CNS depressant drugs or alcohol, and caution that the effects of other CNS depressant drugs or alcohol may be potentiated [see Warnings and Precautions (5.4 )] .
Effect of ONFI on Other Drugs
Hormonal Contraceptives ONFI is a weak CYP3A4 inducer. As some hormonal contraceptives are metabolized by CYP3A4, their effectiveness may be diminished when given with ONFI. Additional non-hormonal forms of contraception are recommended when using ONFI [see Clinical Pharmacology (12.3 ), Patient Counseling Information (17 )] .
Drugs Metabolized by CYP2D6 ONFI inhibits CYP2D6. Dose adjustment of drugs metabolized by CYP2D6 may be necessary [see Clinical Pharmacology (12.3 )] .
Effect of Other Drugs on ONFI
Strong and moderate inhibitors of CYP2C19 Strong and moderate inhibitors of CYP2C19 may result in increased exposure to N-desmethylclobazam, the active metabolite of clobazam. This may increase the risk of dose-related adverse reactions. Dosage adjustment of ONFI may be necessary when co-administered with strong CYP2C19 inhibitors (e.g., fluconazole, fluvoxamine, ticlopidine) or moderate CYP2C19 inhibitors (e.g., omeprazole) [see Clinical Pharmacology (12.3 )] .
11 DESCRIPTION
| Proprietary Name: | ONFI ® |
| Established Name: | Clobazam |
| Dosage Forms: | Tablet and Oral Suspension |
| Route of Administration: | Oral |
| Established Pharmacologic Class of Drug: | Benzodiazepine |
| Chemical Name: | 7-Chloro-1-methyl-5-phenyl-1H-1,5 benzodiazepine-2,4( 3H,5H )-dione |
| Structural Formula: |
|
Clobazam is a white or almost white, crystalline powder with a slightly bitter taste; is slightly soluble in water, sparingly soluble in ethanol, and freely soluble in methylene chloride. The melting range of clobazam is from 182ºC to 185ºC. The molecular formula is C 16 H 13 O 2 N 2 Cl and the molecular weight is 300.7.
Each ONFI tablet contains 10 mg or 20 mg of clobazam. Tablets also contain as inactive ingredients: modified corn starch, lactose monohydrate, magnesium stearate, silicon dioxide, and talc.
ONFI is also available for oral administration as an off-white suspension containing clobazam at a concentration of 2.5 mg/mL. Inactive ingredients include magnesium aluminum silicate, xanthan gum, citric acid monohydrate, disodium hydrogen phosphate dihydrate, simethicone emulsion, polysorbate 80, methylparaben, propylparaben, propylene glycol, sucralose, maltitol solution, berry flavor, purified water.
12 CLINICAL PHARMACOLOGY
Mechanism of Action
The exact mechanism of action for clobazam, a 1,5-benzodiazepine, is not fully understood but is thought to involve potentiation of GABAergic neurotransmission resulting from binding at the benzodiazepine site of the GABA A receptor.
Pharmacodynamics
Effects on Electrocardiogram The effect of ONFI 20 mg and 80 mg administered twice daily on QTc interval was evaluated in a randomized, evaluator-blinded, placebo-, and active-controlled (moxifloxacin 400 mg) parallel thorough QT study in 280 healthy subjects. In a study with demonstrated ability to detect small effects, the upper bound of the one-sided 95% confidence interval for the largest placebo-adjusted, baseline-corrected QTc based on the Fridericia correction method was below 10 ms, the threshold for regulatory concern. Thus, at a dose two times the maximum recommended dose, ONFI did not prolong the QTc interval to any clinically relevant extent.
Pharmacokinetics
The peak plasma levels (C max ) and the area under the curve (AUC) of clobazam are dose-proportional over the dose range of 10-80 mg following single- or multiple-dose administration of ONFI. Based on a population pharmacokinetic analysis, the pharmacokinetics of clobazam are linear from 5-160 mg/day. Clobazam is converted to N-desmethylclobazam which has about 1/5 the activity of clobazam. The estimated mean elimination half-lives (t 1/2 ) of clobazam and N-desmethylclobazam were 36-42 hours and 71-82 hours, respectively.
Absorption Clobazam is rapidly and extensively absorbed following oral administration. The time to peak concentrations (T max ) of clobazam tablets under fasted conditions ranged from 0.5 to 4 hours after single- or multiple-dose administrations. The relative bioavailability of clobazam tablets compared to an oral solution is approximately 100%. After single dose administration of the oral suspension under fasted conditions, the T max ranged from 0.5 to 2 hours. Based on exposure (C max and AUC) of clobazam, ONFI tablets and suspension were shown to have similar bioavailability under fasted conditions. The administration of ONFI tablets with food or when crushed in applesauce does not affect absorption. Although not studied, the oral bioavailability of the oral suspension is unlikely to be affected under fed conditions.
Distribution Clobazam is lipophilic and distributes rapidly throughout the body. The apparent volume of distribution at steady state was approximately 100 L. The in vitro plasma protein binding of clobazam and N-desmethylclobazam is approximately 80-90% and 70%, respectively.
Metabolism and Excretion Clobazam is extensively metabolized in the liver, with approximately 2% of the dose recovered in urine and 1% in feces as unchanged drug. The major metabolic pathway of clobazam involves N-demethylation, primarily by CYP3A4 and to a lesser extent by CYP2C19 and CYP2B6. N-desmethylclobazam, an active metabolite, is the major circulating metabolite in humans, and at therapeutic doses, plasma concentrations are 3-5 times higher than those of the parent compound. Based on animal and in vitro receptor binding data, estimates of the relative potency of N-desmethylclobazam compared to parent compound range from 1/5 to equal potency. N-desmethylclobazam is extensively metabolized, mainly by CYP2C19. N-desmethylclobazam and its metabolites comprise ~94% of the total drug-related components in urine. Following a single oral dose of radiolabeled drug, approximately 11% of the dose was excreted in the feces and approximately 82% was excreted in the urine.
The polymorphic CYP2C19 is the major contributor to the metabolism of the pharmacologically active N-desmethylclobazam [see Clinical Pharmacology (12.5 )] . In CYP2C19 poor metabolizers, levels of N-desmethylclobazam were 5-fold higher in plasma and 2- to 3-fold higher in the urine than in CYP2C19 extensive metabolizers.
Pharmacokinetics in Specific Populations Age Population pharmacokinetic analyses showed that the clearance of clobazam is lower in elderly subjects compared to other age groups (ages 2 to 64). Dosing should be adjusted in the elderly [see Dosage and Administration (2.4 )] .
Sex Population pharmacokinetic analyses showed no difference in the clearance of clobazam between women and men.
Race Population pharmacokinetic analyses including Caucasian (75%), African American (15%), and Asian (9%) subjects showed that there is no evidence of clinically significant effect of race on the clearance of clobazam.
Renal Impairment The effect of renal impairment on the pharmacokinetics of clobazam was evaluated in patients with mild (creatinine clearance [CL CR ] >50 to 80 mL/min; N=6) and moderate (CL CR =30 to 50 mL/min; N=6) renal dysfunction, with matching healthy controls (N=6), following administration of multiple doses of ONFI 20 mg/day. There were insignificant changes in C max (3-24%) and AUC (≤13%) for clobazam or N-desmethylclobazam in patients with mild or moderate renal impairment compared to patients with normal renal function. Patients with severe renal impairment or ESRD were not included in this study.
Hepatic Impairment There are limited data to characterize the effect of hepatic impairment on the pharmacokinetics of clobazam. In a small study, the pharmacokinetics of a 20 mg single oral dose of ONFI in 9 patients with liver impairment were compared to healthy controls (N=6). The C max and the mean plasma clearance of clobazam, as well as the C max of N-desmethylclobazam, showed no significant change compared to the healthy controls. The AUC values of N-desmethylclobazam in these patients were not available. Adjust dosage in patients with hepatic impairment [see Dosage and Administration (2.7 )] .
Drug Interaction Studies
In vitro studies : Clobazam did not inhibit CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, UGT1A1, UGT1A4, UGT1A6, or UGT2B4 in vitro . N-desmethylclobazam showed weak inhibition of CYP2C9, UGT1A4, UGT1A6 and UGT2B4.
Clobazam and N-desmethylclobazam did not significantly increase CYP1A2 or CYP2C19 activities, but did induce CYP3A4 activity in a concentration-dependent manner. Clobazam and N-desmethylclobazam also increased UGT1A1 mRNA but at concentrations much higher than therapeutic levels. The potential for clobazam or N-desmethylclobazam to induce CYP2B6 and CYP2C8 has not been evaluated.
Clobazam and N-desmethylclobazam do not inhibit P-glycoprotein (P-gp), but are P-gp substrates.
In vivo studies :
Potential for ONFI to Affect Other Drugs The effect of repeated 40 mg once-daily doses of ONFI on the pharmacokinetic profiles of single-dose dextromethorphan (CYP2D6 substrate), midazolam (CYP3A4 substrate), caffeine (CYP1A2 substrate), and tolbutamide (CYP2C9 substrate), was studied when these probe substrates were given as a drug cocktail (N=18).
Clobazam increased AUC and C max of dextromethorphan by 90% and 59%, respectively, reflecting its inhibition of CYP2D6 in vivo . Drugs metabolized by CYP2D6 may require dose adjustment when used with ONFI.
Clobazam decreased the AUC and C max of midazolam by 27% and 24%, respectively, and increased the AUC and C max of the metabolite 1-hydroxymidazolam by 4-fold and 2-fold, respectively. This level of induction does not call for dosage adjustment of drugs that are primarily metabolized by CYP3A4 when used concomitantly with ONFI. Some hormonal contraceptives are metabolized by CYP3A4 and their effectiveness may be diminished when given with ONFI [see Drug Interactions (7.3 )] . Repeated ONFI doses had no effect on caffeine and tolbutamide.
A population pharmacokinetic analysis indicated clobazam did not affect the exposure of valproic acid (a CYP2C9/2C19 substrate) or lamotrigine (a UGT substrate).
Potential for Other Drugs to Affect ONFI Co-administration of ketoconazole (a strong CYP3A4 inhibitor) 400 mg once-daily for 5 days increased clobazam AUC by 54%, with an insignificant effect on clobazam C max . There was no significant change in AUC and C max of N-desmethylclobazam (N=18).
Strong (e.g., fluconazole, fluvoxamine, ticlopidine) and moderate (e.g., omeprazole) inhibitors of CYP2C19 may result in up to a 5-fold increase in exposure to N-desmethylclobazam, the active metabolite of clobazam, based on extrapolation from pharmacogenomic data [see Clinical Pharmacology (12.5 )] . Dosage adjustment of ONFI may be necessary when co-administered with strong or moderate CYP2C19 inhibitors [see Drug Interactions (7.4 )] .
The effects of concomitant antiepileptic drugs that are CYP3A4 inducers (phenobarbital, phenytoin, and carbamazepine), CYP2C19 inducers (valproic acid, phenobarbital, phenytoin, and carbamazepine), and CYP2C19 inhibitors (felbamate and oxcarbazepine) were evaluated using data from clinical trials. Results of population pharmacokinetic analysis show that these concomitant antiepileptic drugs did not significantly alter the pharmacokinetics of clobazam or N-desmethylclobazam at steady-state.
Alcohol has been reported to increase the maximum plasma exposure of clobazam by approximately 50%. Alcohol may have additive CNS depressant effects when taken with ONFI [see Warnings and Precautions (5.4 ), Drug Interactions (7.2 )] .
Pharmacogenomics
The polymorphic CYP2C19 is the main enzyme that metabolizes the pharmacologically active N-desmethylclobazam. Compared to CYP2C19 extensive metabolizers, N-desmethylclobazam AUC and C max are approximately 3-5 times higher in poor metabolizers (e.g., subjects with •2/•2 genotype) and 2 times higher in intermediate metabolizers (e.g., subjects with •1/•2 genotype). The prevalence of CYP2C19 poor metabolism differs depending on racial/ethnic background. Dosage in patients who are known CYP2C19 poor metabolizers may need to be adjusted [see Dosage and Administration (2.5 )] .
The systemic exposure of clobazam is similar for both CYP2C19 poor and extensive metabolizers.
13 NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis In mice, oral administration of clobazam (0, 6, 12, or 24 mg/kg/day) for 2 years did not result in an increase in tumors. The highest dose tested was approximately 3 times the maximum recommended human dose (MRHD) of 40 mg/day, based on body surface area (mg/m 2 ).
In rats, oral administration of clobazam for 2 years resulted in increases in tumors of the thyroid gland (follicular cell adenoma and carcinoma) and liver (hepatocellular adenoma) at the mid and high doses. The low dose, not associated with an increase in tumors, was associated with plasma exposures (AUC) for clobazam and its major active metabolite, N-desmethylclobazam, less than that in humans at the MRHD.
Mutagenesis Clobazam and the major active metabolite, N-desmethylclobazam, were negative for genotoxicity, based on data from a battery of in vitro (bacteria reverse mutation, mammalian clastogenicity) and in vivo (mouse micronucleus) assays.
Impairment of Fertility In a fertility study in which clobazam (50, 350, or 750 mg/kg/day, corresponding to 12, 84 and 181 times the oral Maximum Recommended Human Dose, MRHD, of 40 mg/day based on mg/m 2 body surface) was orally administered to male and female rats prior to and during mating and continuing in females to gestation day 6, increases in abnormal sperm and pre-implantation loss were observed at the highest dose tested. The no-effect level for fertility and early embryonic development in rats was associated with plasma exposures (AUC) for clobazam and its major active metabolite, N-desmethylclobazam, less than those in humans at the maximum recommended human dose of 40 mg/day.
14 CLINICAL STUDIES
The effectiveness of ONFI for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome was established in two multicenter controlled studies (Study 1 and Study 2). Both studies were similar in terms of disease characteristics and concomitant AED treatments. The most common concomitant AED treatments at baseline included: valproate, lamotrigine, levetiracetam, and topiramate.
Study 1 Study 1 (N=238) was a randomized, double-blind, placebo-controlled study consisting of a 4-week baseline period followed by a 3-week titration period and 12-week maintenance period. Patients age 2-54 years with a current or prior diagnosis of LGS were stratified into 2 weight groups (12.5 kg to ≤30 kg or >30 kg) and then randomized to placebo or one of three target maintenance doses of ONFI according to Table 5.
| ≤30 kg Body Weight | >30 kg Body Weight | |
| Low Dose | 5 mg daily | 10 mg daily |
| Medium Dose | 10 mg daily | 20 mg daily |
| High Dose | 20 mg daily | 40 mg daily |
Doses above 5 mg/day were administered in two divided doses.
The primary efficacy measure was the percent reduction in the weekly frequency of drop seizures (atonic, tonic, or myoclonic), also known as drop attacks, from the 4-week baseline period to 12-week maintenance period.
The pre-dosing baseline mean weekly drop seizure frequency was 98, 100, 61, and 105 for the placebo, low-, medium-, and high-dose groups, respectively. Figure 1 presents the mean percent reduction in weekly drop seizures from this baseline. All dose groups of ONFI were statistically superior (p≤0.05) to the placebo group. This effect appeared to be dose dependent.
Figure 1. Mean Percent Reduction from Baseline in Weekly Drop Seizure Frequency (Study 1) 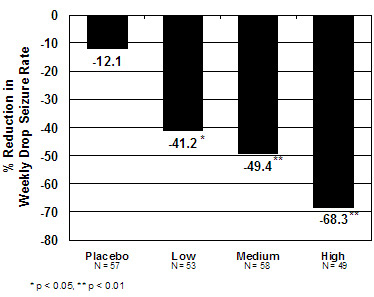
Figure 2 shows changes from baseline in weekly drop seizure frequency by category for patients treated with ONFI and placebo in Study 1. Patients in whom the seizure frequency increased are shown at left as "worse." Patients in whom the seizure frequency decreased are shown in five categories.
Figure 2. Drop Seizure Response by Category for ONFI and Placebo (Study 1) 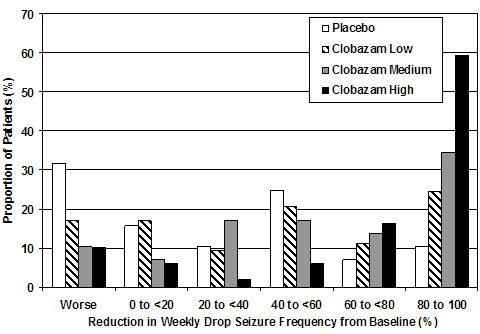
There was no evidence that tolerance to the therapeutic effect of ONFI developed during the 3-month maintenance period.
Study 2 Study 2 (N=68) was a randomized, double-blind comparison study of high- and low-dose ONFI, consisting of a 4-week baseline period followed by a 3-week titration period and 4-week maintenance period. Patients age 2-25 years with a current or prior diagnosis of LGS were stratified by weight, then randomized to either a low or high dose of ONFI, and then entered a 3-week titration period.
The primary efficacy measure was the percent reduction in the weekly frequency of drop seizures (atonic, tonic, or myoclonic), also known as drop attacks, from the 4-week baseline period to the 4-week maintenance period.
A statistically significantly greater reduction in seizure frequency was observed in the high-dose group compared to the low-dose group (median percent reduction of 93% vs 29%; p<0.05).
16 HOW SUPPLIED/STORAGE AND HANDLING
Each ONFI tablet contains 10 mg or 20 mg of clobazam and is a white to off-white, oval tablet with a functional score on one side and either a "1" and "0" or a "2" and "0" debossed on the other side.
NDC 67386-314-01: 10 mg scored tablet, Bottles of 100 NDC 67386-315-01: 20 mg scored tablet, Bottles of 100
ONFI oral suspension is a berry flavored off-white liquid supplied in a bottle with child-resistant closure. The oral suspension is packaged with a dispenser set which contains two calibrated oral dosing syringes and a bottle adapter.
Store and dispense ONFI oral suspension in its original bottle in an upright position. Use within 90 days of first opening the bottle, then discard any remainder.
NDC 67386-313-21: 2.5 mg/mL supplied in a bottle containing 120 mL of suspension.
Store tablets and oral suspension at 20°C to 25°C (68°F to 77°F). See USP controlled room temperature.
Mechanism of Action
The exact mechanism of action for clobazam, a 1,5-benzodiazepine, is not fully understood but is thought to involve potentiation of GABAergic neurotransmission resulting from binding at the benzodiazepine site of the GABA A receptor.