Get your patient on Prezcobix (Darunavir Ethanolate And Cobicistat)
Prezcobix patient education
Patient toolkit
Dosage & administration

Prezcobix prescribing information
INDICATIONS AND USAGE
PREZCOBIX and PREZCOBIX PED are indicated in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV-1) in treatment-naïve and treatment-experienced adults and pediatric patients 3 years of age and older weighing at least 15 kg with no darunavir resistance-associated substitutions (V11I, V32I, L33F, I47V, I50V, I54L, I54M, T74P, L76V, I84V, L89V) [see Use in Specific Populations (8.4) and Clinical Studies (14) ] .
DOSAGE AND ADMINISTRATION
- Adults and pediatric patients weighing at least 40 kg: One 800 mg/150 mg tablet taken once daily with food. (2.1 , 2.2 )
- Pediatric patients weighing at least 25 kg to less than 40 kg: One 675 mg/150 mg tablet taken once daily with food. (2.1 , 2.2 )
- Pediatric patients 3 years of age and older weighing at least 15 kg to less than 25 kg: One 600 mg/90 mg tablet for oral suspension taken once daily with food. PREZCOBIX PED must be dispersed in drinking water and taken immediately with food. (2.2 , 2.3 )
Testing Prior to Initiation: HIV genotypic testing is recommended for antiretroviral treatment experienced patients. Assess estimated creatinine clearance in all patients prior to starting PREZCOBIX or PREZCOBIX PED. When used with tenofovir disoproxil fumarate (TDF): Assess urine glucose and urine protein at baseline and monitor creatinine clearance, urine glucose, and urine protein. Monitor serum phosphorus in patients with or at risk for renal impairment. (2.5 )
Overview of Different Dosage Forms
Two different dosage forms are available:
- PREZCOBIX Tablets:
- 800 mg/150 mg film-coated tablets for adults and pediatric patients weighing at least 40 kg.
- 675 mg/150 mg film-coated tablets for pediatric patients weighing at least 25 kg to less than 40 kg.
- PREZCOBIX PED Tablets for Oral Suspension:
- 600 mg/90 mg film-coated tablet for oral suspension for pediatric patients aged 3 years and older weighing at least 15 kg to less than 25 kg [see Dosage and Administration (2.2 , 2.3) ] .
Recommended Dosage in Adults and Pediatrics 3 Years of Age and Older Weighing at Least 15 kg
The recommended dosages for adults and pediatric patients weighing at least 15 kg are shown in Table 1. The pediatric dose is based on weight. PREZCOBIX and PREZCOBIX PED are taken orally with food once daily in conjunction with other antiretroviral agents.
| Patient Population | Total Daily Dose |
|---|---|
| Adults and pediatric patients weighing at least 40 kg | One PREZCOBIX 800 mg darunavir/150 mg cobicistat tablet |
| Pediatric Patients weighing at least 25 kg to less than 40 kg | One PREZCOBIX 675 mg darunavir/150 mg cobicistat tablet |
| Pediatric Patients aged 3 years and older weighing at least 15 kg to less than 25 kg | One PREZCOBIX PED 600 mg darunavir/90 mg cobicistat tablet for oral suspension |
Before prescribing PREZCOBIX 675 mg/150 mg tablets, children should be assessed for the ability to swallow tablets. For patients unable to swallow the 675 mg/150 mg tablet whole, the scored tablet may be split by hand into two pieces. Each piece should be consumed immediately after splitting to ensure the entire dose is administered. The score line is only to facilitate breaking for ease of swallowing and not to divide into equal doses.
Preparation and Administration Instructions for PREZCOBIX PED
Advise patients or caregivers of patients taking PREZCOBIX PED to refer to the Instructions for Use to properly prepare and take the medication.
PREZCOBIX PED must be dispersed in drinking water and taken immediately as described below. If not taken immediately, then the oral suspension should be discarded, and a new dose of medicine should be prepared. The patient should not crush, chew or swallow the PREZCOBIX PED tablet for oral suspension. The following instructions should be followed:
- Place the tablet for oral suspension in a cup, add 30 mL (2 tablespoons) of non-carbonated room temperature drinking water.
- Stir well with a spoon until the tablet is completely dispersed. The prepared medicine will appear reddish purple.
- Take all the prepared medicine immediately or to aid in administration, the prepared medicine can be further diluted with 10 mL (2 teaspoons) of water, orange juice, or 1 teaspoon of applesauce or yogurt. Mix and take the entire mixture immediately.
- Add another 5 mL (1 teaspoon) of non-carbonated drinking water to the cup, swirl and drink completely. This step may be repeated as needed to ensure the entire dose is consumed.
Testing Prior to Initiation of PREZCOBIX or PREZCOBIX PED
HIV genotypic testing is recommended for antiretroviral treatment-experienced patients. However, when HIV genotypic testing is not feasible, PREZCOBIX or PREZCOBIX PED can be used in protease inhibitor-naïve patients, but is not recommended in protease inhibitor-experienced patients.
Creatinine ClearancePrior to starting PREZCOBIX or PREZCOBIX PED, assess estimated creatinine clearance because cobicistat decreases estimated creatinine clearance due to inhibition of tubular secretion of creatinine without affecting actual renal glomerular function [see Warnings and Precautions (5.3) ] . When co-administering PREZCOBIX or PREZCOBIX PED with tenofovir disoproxil fumarate (tenofovir DF) assess estimated creatinine clearance, urine glucose, and urine protein at baseline [see Warnings and Precautions (5.4) ] .
Not Recommended in Severe Renal Impairment
PREZCOBIX or PREZCOBIX PED co-administered with tenofovir DF is not recommended in patients who have an estimated creatinine clearance below 70 mL per minute [see Warnings and Precautions (5.4) and Adverse Reactions (6) ] .
Not Recommended in Severe Hepatic Impairment
PREZCOBIX or PREZCOBIX PED is not recommended for use in patients with severe hepatic impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3) ] .
Not Recommended During Pregnancy
PREZCOBIX or PREZCOBIX PED is not recommended during pregnancy because of substantially lower exposures of darunavir and cobicistat during the second and third trimesters [see Use in Specific Populations (8.1) and Clinical Pharmacology (12.3) ] .
PREZCOBIX or PREZCOBIX PED should not be initiated in pregnant individuals. An alternative regimen is recommended for those who become pregnant during therapy with PREZCOBIX or PREZCOBIX PED.
DOSAGE FORMS AND STRENGTHS
- 800 mg/150 mg as pink, oval-shaped, film-coated tablet debossed with "800" on one side and "TG" on the other side containing 800 mg of darunavir and 150 mg of cobicistat.
- 675 mg/150 mg as green to dark green, oval-shaped, scored film-coated tablet debossed with "675" on one side and "TG" on the other side containing 675 mg of darunavir and 150 mg of cobicistat.
- 600 mg/90 mg as reddish purple, oval-shaped, film-coated tablet debossed with "690" on one side and "TG" on the other side containing 600 mg of darunavir and 90 mg of cobicistat.
USE IN SPECIFIC POPULATIONS
- Pregnancy: PREZCOBIX or PREZCOBIX PED is not recommended during pregnancy due to substantially lower exposures of darunavir and cobicistat during pregnancy. (8.1 , 12.3 )
- Lactation: Breastfeeding is not recommended. (8.2 )
- Pediatrics: Not recommended for pediatric patients younger than 3 years or weighing less than 15 kg. (8.4 )
Pregnancy
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to PREZCOBIX or PREZCOBIX PED during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) 1-800-258-4263.
Risk SummaryPREZCOBIX or PREZCOBIX PED is not recommended during pregnancy because of substantially lower exposures of darunavir and cobicistat during the second and third trimesters [see Dosage and Administration (2.7) ] . A study evaluating the pharmacokinetics of antiretrovirals during pregnancy demonstrated substantially lower exposures of darunavir and cobicistat in the second and third trimesters compared to the post-partum period (see Data ) and [see Clinical Pharmacology (12.3) ].
Prospective pregnancy data from the APR are not sufficient to adequately assess the risk of birth defects or miscarriage. However, available data from the APR show no statistically significant difference in the overall risk of major birth defects for darunavir and cobicistat compared with the background rate for major birth defects of 2.7% in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data ) . The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15–20%. The background risk of major birth defects and miscarriage for the indicated population is unknown.
In animal reproduction studies, no adverse developmental effects were observed when the components of PREZCOBIX were administered separately at darunavir exposures less than 1 (mice and rabbits) and 3-times (rats), and at cobicistat exposures 1.6 (rats) and 3.8 (rabbits) times human exposures at the recommended daily dose of these components in PREZCOBIX (see Data ) . No adverse developmental effects were seen when cobicistat was administered to rats through lactation at cobicistat exposures up to 1.2 times the human exposure at the recommended therapeutic dose.
Clinical Considerations Not Recommended During PregnancyPREZCOBIX or PREZCOBIX PED is not recommended for use during pregnancy because of substantially lower exposures of darunavir and cobicistat during pregnancy (see Data ) and [see Clinical Pharmacology (12.3) ] .
PREZCOBIX or PREZCOBIX PED should not be initiated in pregnant individuals. An alternative regimen is recommended for individuals who become pregnant during therapy with PREZCOBIX or PREZCOBIX PED.
Data Human DataPREZCOBIX in combination with a background regimen was evaluated in a clinical trial of 7 pregnant participants taking PREZCOBIX (800 mg/150 mg) prior to enrollment and who were willing to remain on PREZCOBIX throughout the study. The study period included the second and third trimesters, and through 12 weeks postpartum. Six pregnant participants completed the trial.
Exposure to darunavir and cobicistat as part of an antiretroviral regimen was substantially lower during the second and third trimesters of pregnancy compared with postpartum [see Clinical Pharmacology (12.3) ] .
One out of 6 pregnant participants who completed the study experienced virologic failure with HIV-1 RNA >1,000 copies/mL from the third trimester visit through the postpartum period. Five pregnant participants had sustained virologic response (HIV-1 RNA <50 copies/mL) throughout the study period. There are no clinical data on the virologic response when PREZCOBIX is initiated during pregnancy.
Prospective reports from the APR of overall major birth defects in pregnancies exposed to the components of PREZCOBIX are compared with a U.S. background major birth defect rate. Methodological limitations of the APR include the use of MACDP as the external comparator group. Limitations of using an external comparator include differences in methodology and populations, as well as confounding due to the underlying disease.
There were no new clinically relevant safety findings compared with the known safety profile of PREZCOBIX in adults with HIV-1.
Darunavir : Based on prospective reports to the APR of over 980 exposures to darunavir-containing regimens during pregnancy resulting in live births (including over 660 exposed in the first trimester and over 320 exposed in the second/third trimester), the prevalence of birth defects in live births was 3.6% (95% CI: 2.3% to 5.3%) with first trimester exposure to darunavir-containing regimens and 2.5% (95% CI: 1.1% to 4.8%) with second/third trimester exposure to darunavir-containing regimens.
Cobicistat : Based on prospective reports to the APR of over 570 exposures to cobicistat-containing regimens during pregnancy resulting in live births (including over 480 exposed in the first trimester and over 80 exposed in the second/third trimester), the prevalence of birth defects in live births was 3.7% (95% CI: 2.2% to 5.7%) and 1.1% (95% CI: 0.0% to 6.2%) with first and second/third trimester, respectively, to cobicistat-containing regimens.
Animal DataDarunavir : Reproduction studies conducted with darunavir showed no embryotoxicity or teratogenicity in mice (doses up to 1000 mg/kg from gestation day (GD) 6–15 with darunavir alone) and rats (doses up to 1000 mg/kg from GD 7–19 in the presence or absence of ritonavir) as well as in rabbits (doses up to 1000 mg/kg/day from GD 8–20 with darunavir alone). In these studies, darunavir exposures (based on AUC) were higher in rats (3-fold), whereas in mice and rabbits, exposures were lower (less than 1-fold) compared to those obtained in humans at the recommended clinical dose of darunavir co-administered with ritonavir.
Cobicistat : Cobicistat was administered orally to pregnant rats at doses up to 125 mg/kg/day on GD 6–17. Increases in post-implantation loss and decreased fetal weights were observed at a maternal toxic dose of 125 mg/kg/day. No malformations were noted at doses up to 125 mg/kg/day. Systemic exposures (AUC) at 50 mg/kg/day in pregnant females were 1.6 times higher than human exposures at the recommended daily dose of cobicistat.
In pregnant rabbits, cobicistat was administered orally at doses up to 100 mg/kg/day during GD 7–20. No maternal or embryo/fetal effects were noted at the highest dose of 100 mg/kg/day. Systemic exposures (AUC) at 100 mg/kg/day were 3.8 times higher than human exposures at the recommended daily dose of cobicistat.
In a pre/postnatal developmental study in rats, cobicistat was administered orally at doses up to 75 mg/kg from GD 6 to postnatal day 20, 21, or 22. At doses of 75 mg/kg/day, neither maternal nor developmental toxicity was noted. Systemic exposures (AUC) at this dose were 1.2 times the human exposures at the recommended daily dose of cobicistat.
Lactation
There are no data on the presence of darunavir or cobicistat in human milk, the effects on the breastfed infant, or the effects on milk production. Darunavir and cobicistat are present in the milk of lactating rats (see Data ) . Potential risks of breastfeeding include: (1) HIV-1 transmission (in infants without HIV-1), (2) developing viral resistance (in infants with HIV-1), and (3) serious adverse reactions in a breastfed infant similar to those seen in adults.
Data Animal DataDarunavir : Studies in rats (with darunavir alone or with ritonavir) have demonstrated that darunavir is excreted in milk. In the rat pre- and postnatal development study, a reduction in pup body weight gain was observed due to exposure of pups to drug substances via milk. The maximal maternal plasma exposures achieved with darunavir (up to 1000 mg/kg with ritonavir) were approximately 50% of those obtained in humans at the recommended clinical dose of darunavir with ritonavir.
Cobicistat : During the pre/postnatal developmental toxicology study at doses up to 75 mg/kg/day, mean cobicistat milk to plasma ratio of up to 1.9 was measured 2 hours after administration to rats on lactation day 10.
Females and Males of Reproductive Potential
Additional or alternative (non-hormonal) forms of contraception should be considered when estrogen-containing contraceptives are co-administered with PREZCOBIX or PREZCOBIX PED. For co-administration with drospirenone, clinical monitoring is recommended due to the potential for hyperkalemia. No data are available to make recommendations on co-administration with other hormonal contraceptives [see Drug Interactions (7.3) ].
Pediatric Use
The safety and effectiveness of PREZCOBIX and PREZCOBIX PED for the treatment of HIV-1 in pediatric patients 3 years of age and older weighing at least 15 kg was established through a trial with components of PREZCOBIX and PREZCOBIX PED. Use of PREZCOBIX or PREZCOBIX PED in this group is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic, safety, and virologic data from a study of components of PREZCOBIX or PREZCOBIX PED (Trial GS-US-216-0128) in pediatric participants with HIV-1 aged 3 to less than 18 years [see Adverse Reactions (6.1) , Clinical Pharmacology (12.3) , and Clinical Studies (14.2) ] .
The safety and effectiveness of PREZCOBIX or PREZCOBIX PED have not been established in pediatric patients weighing less than 15 kg. Darunavir, a component of PREZCOBIX and PREZCOBIX PED is not recommended in pediatric patients below 3 years of age because of toxicity and mortality observed in juvenile rats dosed with darunavir.
Juvenile Animal Toxicity Data
Darunavir: In a juvenile toxicity study where rats were directly dosed with darunavir (up to 1000 mg/kg), deaths occurred from post-natal day 5 at plasma exposure levels ranging from 0.1 to 1.0 of the human exposure levels. In a 4-week rat toxicology study, when dosing was initiated on post-natal day 23 (the human equivalent of 2 to 3 years of age), no deaths were observed with a plasma exposure (in combination with ritonavir) 2 times the human plasma exposure levels.
Geriatric Use
Clinical trials of PREZCOBIX did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. In general, caution should be exercised in the administration and monitoring of PREZCOBIX in elderly patients, reflecting the greater frequency of decreased hepatic function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3) ] .
Hepatic Impairment
No clinical trials were conducted with darunavir co-administered with cobicistat in hepatically impaired patients and the effect of hepatic impairment on darunavir exposure when co-administered with cobicistat has not been evaluated. Based on the recommendations for darunavir co-administered with ritonavir, a dose adjustment for patients with mild or moderate hepatic impairment is not necessary. No pharmacokinetic or safety data are available regarding the use of darunavir in patients with severe hepatic impairment. Therefore, PREZCOBIX or PREZCOBIX PED is not recommended for use in patients with severe hepatic impairment [see Clinical Pharmacology (12.3) ] .
Renal Impairment
A renal impairment trial was not conducted for darunavir co-administered with cobicistat [see Clinical Pharmacology (12.3) ] . Cobicistat has been shown to decrease estimated creatinine clearance without affecting actual renal glomerular function. Dosing recommendations are not available for drugs that require dosage adjustment for renal impairment when used in combination with PREZCOBIX or PREZCOBIX PED [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.2) ] .
CONTRAINDICATIONS
Darunavir and cobicistat are both inhibitors of the cytochrome P450 3A (CYP3A) isoform. PREZCOBIX should not be co-administered with medicinal products that are highly dependent on CYP3A for clearance and for which increased plasma concentrations are associated with serious and/or life-threatening events (narrow therapeutic index). Darunavir and cobicistat are both substrates of the cytochrome P450 3A (CYP3A) isoform. Co-administration of PREZCOBIX or PREZCOBIX PED with CYP3A inducers may lead to lower exposures of darunavir and cobicistat and potential loss of efficacy of darunavir and possible resistance. Examples of drugs that are contraindicated for co-administration with PREZCOBIX or PREZCOBIX PED [see Drug Interactions (7.3 ) and Clinical Pharmacology (12.3) ] are listed below.
- Alpha 1-adrenoreceptor antagonist: alfuzosin
- Anticonvulsants: carbamazepine, phenobarbital, phenytoin
- Anti-gout: colchicine, in patients with renal and/or hepatic impairment
- Antimycobacterial: rifampin
- Antipsychotics: lurasidone, pimozide
- Cardiac Disorders: dronedarone, ivabradine, ranolazine
- Ergot derivatives, e.g. dihydroergotamine, ergotamine, methylergonovine
- Herbal product: St. John's wort ( Hypericum perforatum )
- Hepatitis C direct acting antiviral: elbasvir/grazoprevir
- Lipid modifying agents: lomitapide, lovastatin, simvastatin
- Opioid Antagonist: naloxegol
- PDE-5 inhibitor: sildenafil when used for treatment of pulmonary arterial hypertension
- Sedatives/hypnotics: orally administered midazolam, triazolam
WARNINGS AND PRECAUTIONS
- Drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis), liver injury, including some fatalities can occur with PREZCOBIX or PREZCOBIX PED. Monitor liver function before and during therapy, especially in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases. (5.1 )
- Skin reactions ranging from mild to severe, including Stevens-Johnson Syndrome, toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms and acute generalized exanthematous pustulosis, can occur with PREZCOBIX or PREZCOBIX PED. Discontinue treatment if severe reaction develops. (5.2 )
- When PREZCOBIX or PREZCOBIX PED is used in combination with a TDF containing regimen, cases of acute renal failure and Fanconi syndrome have been reported. (5.4 )
- PREZCOBIX or PREZCOBIX PED is not recommended in combination with other antiretroviral drugs that require pharmacokinetic boosting. (5.6 )
- Monitor in patients with a known sulfonamide allergy. (5.7 )
- Patients receiving PREZCOBIX or PREZCOBIX PED may develop new onset or exacerbations of diabetes mellitus/hyperglycemia (5.8 ), redistribution/accumulation of body fat (5.9 ), and immune reconstitution syndrome. (5.10 )
- Patients with hemophilia may develop increased bleeding events. (5.11 )
Hepatotoxicity
During the darunavir clinical development program (N=3063), where darunavir was co-administered with ritonavir 100 mg once or twice daily, drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis) was reported in 0.5% of participants. Patients with pre-existing liver dysfunction, including chronic active hepatitis B or C, have an increased risk for liver function abnormalities including severe hepatic adverse reactions.
Post-marketing cases of liver injury, including some fatalities, have also been reported with darunavir co-administered with ritonavir. These have generally occurred in patients with advanced HIV-1 disease taking multiple concomitant medications, having co-morbidities including hepatitis B or C co-infection, and/or developing immune reconstitution syndrome. A causal relationship with darunavir co-administered with ritonavir has not been established.
Appropriate laboratory testing should be conducted prior to initiating therapy with PREZCOBIX or PREZCOBIX PED and patients should be monitored during treatment. Increased AST/ALT monitoring should be considered in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases, especially during the first several months of PREZCOBIX or PREZCOBIX PED treatment.
Evidence of new or worsening liver dysfunction (including clinically significant elevation of liver enzymes and/or symptoms such as fatigue, anorexia, nausea, jaundice, dark urine, liver tenderness, hepatomegaly) in patients on PREZCOBIX or PREZCOBIX PED should prompt consideration of interruption or discontinuation of treatment.
Severe Skin Reactions
During the darunavir clinical development program (n=3063), where darunavir was co-administered with ritonavir 100 mg once or twice daily, severe skin reactions, accompanied by fever and/or elevations of transaminases in some cases, was reported in 0.4% of participants. Stevens-Johnson Syndrome was rarely (less than 0.1%) reported during the clinical development program. During post-marketing experience toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms, and acute generalized exanthematous pustulosis have been reported. Discontinue PREZCOBIX or PREZCOBIX PED immediately if signs or symptoms of severe skin reactions develop. These can include but are not limited to severe rash or rash accompanied with fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, hepatitis and/or eosinophilia.
Mild-to-moderate rash was also reported and often occurred within the first four weeks of treatment and resolved with continued dosing.
Effects on Serum Creatinine
Cobicistat decreases estimated creatinine clearance due to inhibition of tubular secretion of creatinine without affecting actual renal glomerular function. This effect should be considered when interpreting changes in estimated creatinine clearance in patients initiating PREZCOBIX or PREZCOBIX PED, particularly in patients with medical conditions or receiving drugs needing monitoring with estimated creatinine clearance.
Prior to initiating therapy with PREZCOBIX or PREZCOBIX PED, assess estimated creatinine clearance [see Dosage and Administration (2.4) ] . Dosage recommendations are not available for drugs that require dosage adjustments in PREZCOBIX or PREZCOBIX PED treated patients with renal impairment [see Drug Interactions (7.3) and Clinical Pharmacology (12.2) ] . Consider alternative medications that do not require dosage adjustments in patients with renal impairment.
Although cobicistat may cause modest increases in serum creatinine and modest declines in estimated creatinine clearance without affecting renal glomerular function, patients who experience a confirmed increase in serum creatinine of greater than 0.4 mg/dL from baseline should be closely monitored for renal safety.
New Onset or Worsening Renal Impairment When Used With Tenofovir Disoproxil Fumarate
Renal impairment, including cases of acute renal failure and Fanconi syndrome, has been reported when cobicistat, a component of PREZCOBIX or PREZCOBIX PED, was used in an antiretroviral regimen that contained tenofovir DF. Co-administration of PREZCOBIX or PREZCOBIX PED and tenofovir DF is not recommended in patients who have an estimated creatinine clearance below 70 mL/min [see Dosage and Administration (2.4) ] .
- Document urine glucose and urine protein at baseline [see Dosage and Administration (2.4) ] and perform routine monitoring of estimated creatinine clearance, urine glucose, and urine protein during treatment when PREZCOBIX or PREZCOBIX PED is used with tenofovir DF. Measure serum phosphorus in patients with or at risk for renal impairment when used with tenofovir DF.
- Co-administration of PREZCOBIX or PREZCOBIX PED and tenofovir DF in combination with concomitant or recent use of a nephrotoxic agent is not recommended.
See cobicistat full prescribing information for additional information regarding cobicistat.
Risk of Serious Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
Initiation of PREZCOBIX or PREZCOBIX PED, which inhibits CYP3A, in patients receiving medications metabolized by CYP3A, or initiation of medications metabolized by CYP3A in patients already receiving PREZCOBIX or PREZCOBIX PED may increase plasma concentrations of medications metabolized by CYP3A and reduce plasma concentrations of active metabolite(s) formed by CYP3A. Initiation of medications that inhibit or induce CYP3A may respectively increase or decrease concentrations of PREZCOBIX or PREZCOBIX PED.
These interactions may lead to:
- clinically significant adverse reactions, potentially leading to severe, life-threatening, or fatal events from higher exposures of concomitant medications.
- clinically significant adverse reactions from higher exposures of PREZCOBIX or PREZCOBIX PED.
- loss of therapeutic effect of the concomitant medications from lower exposures of active metabolite(s).
- loss of therapeutic effect of PREZCOBIX or PREZCOBIX PED and possible development of resistance from lower exposures of PREZCOBIX or PREZCOBIX PED.
See Table 2 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during PREZCOBIX or PREZCOBIX PED therapy; review concomitant medications during PREZCOBIX or PREZCOBIX PED therapy; and monitor for the adverse reactions associated with concomitant medications [see Contraindications (4) and Drug Interactions (7) ] .
When used with concomitant medications, PREZCOBIX or PREZCOBIX PED may result in different drug interactions than those observed or expected with darunavir co-administered with ritonavir. Complex or unknown mechanisms of drug interactions preclude extrapolation of drug interactions with darunavir co-administered with ritonavir to certain PREZCOBIX or PREZCOBIX PED interactions [see Drug Interactions (7) and Clinical Pharmacology (12.3) ] .
Antiretrovirals Not Recommended
PREZCOBIX or PREZCOBIX PED is not recommended in combination with other antiretroviral drugs that require pharmacokinetic boosting (i.e., another protease inhibitor or elvitegravir) because dosing recommendations for such combinations have not been established and co-administration may result in decreased plasma concentrations of the antiretroviral agents, leading to loss of therapeutic effect and development of resistance.
PREZCOBIX or PREZCOBIX PED is not recommended in combination with products containing the individual components of PREZCOBIX or PREZCOBIX PED (darunavir and cobicistat) or with ritonavir. For additional recommendations on use of PREZCOBIX or PREZCOBIX PED with other antiretroviral agents, [see Drug Interactions (7) ] .
Sulfa Allergy
Darunavir contains a sulfonamide moiety. Monitor patients with a known sulfonamide allergy after initiating PREZCOBIX or PREZCOBIX PED. In clinical studies with darunavir co-administered with ritonavir, the incidence and severity of rash were similar in participants with or without a history of sulfonamide allergy.
Diabetes Mellitus/Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in patients with HIV-1 receiving HIV protease inhibitor (PI) therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued PI therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and causal relationships between HIV PI therapy and these events have not been established.
Fat Redistribution
Redistribution/accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including PREZCOBIX. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of antiretroviral treatment.
Hemophilia
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis in patients with hemophilia type A and B treated with HIV PIs. In some patients, additional factor VIII was given. In more than half of the reported cases, treatment with HIV PIs was continued or reintroduced if treatment had been discontinued. A causal relationship between PI therapy and these episodes has not been established.
ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1) ]
- Severe skin reactions [see Warnings and Precautions (5.2) ]
- Effects on serum creatinine [see Warnings and Precautions (5.3) ]
- New onset or worsening renal impairment when used with tenofovir DF [see Warnings and Precautions (5.4) ]
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.10) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Clinical Trials in AdultsDuring the darunavir clinical development program, where darunavir was co-administered with ritonavir 100 mg once or twice daily, the most common clinical adverse reactions (incidence greater than or equal to 5%) of at least moderate intensity (greater than or equal to Grade 2) were diarrhea, nausea, rash, headache, abdominal pain, and vomiting. See the darunavir full prescribing information for additional information on adverse reactions reported with darunavir co-administered with ritonavir. See cobicistat full prescribing information for clinical trial information on adverse reactions reported with cobicistat.
One single arm clinical trial was conducted with darunavir and cobicistat administered as single entities in 313 participants with HIV-1. Adverse reactions evaluated through Week 24 did not differ substantially from those reported in clinical trials with darunavir co-administered with ritonavir.
Clinical Trials in PediatricsNo clinical trials with PREZCOBIX and PREZCOBIX PED were performed in pediatric participants. However, the safety of the components of PREZCOBIX, darunavir and cobicistat, co-administered with two nucleoside reverse transcriptase inhibitors, was evaluated through clinical trial GS-US-216-0128 in virologically-suppressed pediatric participants of 12 to less than 18 years of age with weight ≥40 kg (Cohort 1, N=7), pediatric participants 6 to less than 12 years of age with weight ≥25 kg to <40 kg (Cohort 2, N=8) and pediatric participants aged ≥3 years with weight ≥15 kg to <25 kg (Cohort 3, N=11) through Week 48. Safety analyses of this trial in these pediatric participants did not identify new safety concerns compared to the known safety profile of PREZCOBIX in adult participants [see Clinical Studies (14.2) ] .
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of darunavir. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Metabolism and Nutrition DisordersRedistribution of body fat
Musculoskeletal and Connective Tissue DisordersRhabdomyolysis (associated with co-administration with HMG-CoA reductase inhibitors)
Renal and Urinary DisordersCrystal nephropathy, crystalluria
Skin and Subcutaneous Tissue DisordersToxic epidermal necrolysis, acute generalized exanthematous pustulosis, drug rash with eosinophilia and systemic symptoms [see Warnings and Precautions (5.2) ] .
DRUG INTERACTIONS
- Co-administration of PREZCOBIX or PREZCOBIX PED with other drugs can alter the concentration of other drugs and other drugs may alter the concentrations of darunavir or cobicistat. Consult the full prescribing information prior to and during treatment for potential drug interactions. (4 , 5.6 , 7 , 12.3 )
Potential for PREZCOBIX or PREZCOBIX PED to Affect Other Drugs
Darunavir co-administered with cobicistat is an inhibitor of CYP3A and CYP2D6. Cobicistat inhibits the following transporters: P-glycoprotein (P-gp), BCRP, MATE1, OATP1B1 and OATP1B3. Therefore, co-administration of PREZCOBIX or PREZCOBIX PED with drugs that are primarily metabolized by CYP3A and/or CYP2D6 or are substrates of P-gp, BCRP, MATE1, OATP1B1 or OATP1B3 may result in increased plasma concentrations of such drugs, which could increase or prolong their therapeutic effect and can be associated with adverse events. Co-administration of PREZCOBIX or PREZCOBIX PED with drugs that have active metabolite(s) formed by CYP3A may result in reduced plasma concentrations of these active metabolite(s), potentially leading to loss of their therapeutic effect (see Table 2 ).
Potential for Other Drugs to Affect PREZCOBIX or PREZCOBIX PED
Darunavir is metabolized by CYP3A. Cobicistat is metabolized by CYP3A, and to a minor extent, by CYP2D6. Co-administration of PREZCOBIX or PREZCOBIX PED and drugs that induce CYP3A activity are expected to increase the clearance of darunavir and cobicistat, resulting in lowered plasma concentrations of darunavir and cobicistat which may lead to loss of therapeutic effect and development of resistance. Co-administration of PREZCOBIX or PREZCOBIX PED and other drugs that inhibit CYP3A may result in increased plasma concentrations of darunavir and cobicistat (see Table 2 ).
Established and Other Potentially Significant Drug Interactions
Table 2 provides dosing recommendations for expected clinically relevant interactions with PREZCOBIX or PREZCOBIX PED (this table is not all inclusive). These recommendations are based on either drug interaction trials or predicted interactions due to the expected magnitude of interaction and potential for serious adverse events or loss of therapeutic effect. The table includes examples of potentially significant interactions but is not all inclusive , and therefore the label of each drug that is co-administered with PREZCOBIX or PREZCOBIX PED should be consulted for information related to the route of metabolism, interaction pathways, potential risks, and specific actions to be taken with regard to co-administration. For the list of examples of contraindicated drugs, [see Contraindications (4) ] .
| Concomitant Drug Class: Drug Name Examples | Effect on Concentration of Darunavir, Cobicistat, or Concomitant Drug | Clinical Comment |
|---|---|---|
| HIV-1 antiviral agents: Nucleoside Reverse Transcriptase Inhibitors (NRTIs) | ||
| didanosine | ↔ darunavir ↔ cobicistat ↔ didanosine | Didanosine should be administered one hour before or two hours after PREZCOBIX or PREZCOBIX PED (administered with food). |
| HIV-1 antiviral agents: Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) | ||
| efavirenz | ↓ cobicistat ↓ darunavir | Co-administration with efavirenz is not recommended because it may result in loss of therapeutic effect and development of resistance to darunavir. |
| etravirine | ↓ cobicistat darunavir: effect unknown | Co-administration with etravirine is not recommended because it may result in loss of therapeutic effect and development of resistance to darunavir. |
| nevirapine | ↓ cobicistat darunavir: effect unknown | Co-administration with nevirapine is not recommended because it may result in loss of therapeutic effect and development of resistance to darunavir. |
| HIV-1 antiviral agents: CCR5 co-receptor antagonists | ||
| maraviroc | ↑ maraviroc | Maraviroc is a substrate of CYP3A. When co-administered with PREZCOBIX or PREZCOBIX PED, patients should receive maraviroc 150 mg twice daily. |
| Other agents | ||
| Alpha 1-adrenoreceptor antagonist: alfuzosin | ↑ alfuzosin | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as hypotension. |
| Antibacterials: clarithromycin, erythromycin, telithromycin | ↑ darunavir ↑ cobicistat ↑ antibacterial | Consider alternative antibiotics with concomitant use of PREZCOBIX or PREZCOBIX PED. |
| Anticancer agents: dasatinib, nilotinib | ↑ anticancer agent | A decrease in the dosage or an adjustment of the dosing interval of dasatinib or nilotinib may be necessary when co-administered with PREZCOBIX or PREZCOBIX PED. Consult the dasatinib and nilotinib prescribing information for dosing instructions. |
| vinblastine, vincristine | For vincristine and vinblastine, consider temporarily withholding the cobicistat-containing antiretroviral regimen in patients who develop significant hematologic or gastrointestinal side effects when PREZCOBIX or PREZCOBIX PED is administered concurrently with vincristine or vinblastine. If the antiretroviral regimen must be withheld for a prolonged period, consider initiating a revised regimen that does not include a CYP3A or P-gp inhibitor. | |
| Anticoagulants: Direct Oral Anticoagulants (DOACs) apixaban | ↑ apixaban | Due to potentially increased bleeding risk, dosing recommendations for co-administration of apixaban with PREZCOBIX or PREZCOBIX PED depend on the apixaban dose. Refer to apixaban dosing instructions for co-administration with P-gp and strong CYP3A inhibitors in apixaban prescribing information. |
| rivaroxaban | ↑ rivaroxaban | Co-administration of rivaroxaban with PREZCOBIX or PREZCOBIX PED is not recommended because it may lead to an increased bleeding risk. |
| dabigatran etexilate edoxaban | ↑ dabigatran ↑ edoxaban | Refer to the dabigatran etexilate or edoxaban prescribing information for recommendations regarding co-administration. The specific recommendations are based on indication, renal function, and effect of the co-administered P-gp inhibitors on the concentration of dabigatran or edoxaban. Clinical monitoring is recommended when a DOAC not affected by CYP3A4 but transported by P-gp, including dabigatran etexilate and edoxaban, is co-administered with PREZCOBIX or PREZCOBIX PED. |
| Other Anticoagulants: | ||
| warfarin | warfarin: effect unknown | Monitor the international normalized ratio (INR) when co-administering with warfarin. |
| Anticonvulsants: carbamazepine, phenobarbital, phenytoin | ↓ darunavir ↓ cobicistat | Co-administration is contraindicated due to potential for reduced plasma concentrations of darunavir, which may result in loss of therapeutic effect and development of resistance. |
| Anticonvulsants with CYP3A induction effects that are NOT contraindicated: e.g. eslicarbazepine, oxcarbazepine | ↓ cobicistat darunavir: effect unknown | Consider alternative anticonvulsant or antiretroviral therapy to avoid potential changes in exposures. If co-administration is necessary, monitor for lack or loss of virologic response. |
| Anticonvulsants that are metabolized by CYP3A: e.g. clonazepam | ↑ clonazepam | Clinical monitoring of anticonvulsants is recommended. |
| Antidepressants: Selective Serotonin Reuptake Inhibitors (SSRIs): e.g. paroxetine, sertraline | SSRIs: effects unknown | When co-administering with SSRIs, TCAs, or trazodone, careful dose titration of the antidepressant to the desired effect, including using the lowest feasible initial or maintenance dose, and monitoring for antidepressant response are recommended. |
| Tricyclic Antidepressants (TCAs): e.g. amitriptyline, desipramine, imipramine, nortriptyline | ↑ TCAs | |
| Other antidepressants: trazodone | ↑ trazodone | |
| Antifungals: itraconazole, isavuconazole, ketoconazole, posaconazole | ↑ darunavir ↑ cobicistat | Monitor for increased darunavir or cobicistat and/or antifungal adverse reactions. |
| ↑ itraconazole ↑ ketoconazole ↑ isavuconazole ↔ posaconazole | Specific dosing recommendations are not available for co-administration with these antifungals. Monitor for increased itraconazole or ketoconazole adverse reactions. | |
| voriconazole | voriconazole: effects unknown | Co-administration with voriconazole is not recommended unless benefit/risk assessment justifies the use of voriconazole. |
| Anti-gout: colchicine | ↑ colchicine | Co-administration is contraindicated in patients with renal and/or hepatic impairment due to potential for serious and/or life-threatening reactions. For patients without renal or hepatic impairment:
|
| Antimalarial: artemether/lumefantrine | artemether: effect unknown lumefantrine: effect unknown | Monitor for a potential decrease of antimalarial efficacy or potential QT prolongation. |
| Antimycobacterials: | ||
| rifampin | ↓ darunavir ↓ cobicistat | Co-administration is contraindicated due to potential for loss of therapeutic effect and development of resistance. |
| rifabutin | ↑ rifabutin cobicistat: effects unknown darunavir: effects unknown | When used in combination with PREZCOBIX or PREZCOBIX PED, the recommended dose of rifabutin is 150 mg every other day. Monitor for rifabutin-associated adverse reactions including neutropenia and uveitis. |
| rifapentine | ↓ darunavir | Co-administration with rifapentine is not recommended. |
| Antipsychotics: lurasidone | ↑ lurasidone | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions. |
| pimozide | ↑ pimozide | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| e.g. perphenazine, risperidone, thioridazine | ↑ antipsychotic | A decrease in the dose of antipsychotics that are metabolized by CYP3A or CYP2D6 may be needed when co-administered with PREZCOBIX or PREZCOBIX PED. |
| quetiapine | ↑ quetiapine | Initiation of PREZCOBIX or PREZCOBIX PED in patients taking quetiapine : Consider alternative antiretroviral therapy to avoid increases in quetiapine exposure. If co-administration is necessary, reduce the quetiapine dose to 1/6 of the current dose and monitor for quetiapine- associated adverse reactions. Refer to the quetiapine prescribing information for recommendations on adverse reaction monitoring. Initiation of quetiapine in patients taking PREZCOBIX or PREZCOBIX PED : Refer to the quetiapine prescribing information for initial dosing and titration of quetiapine. |
| β-Blockers: e.g. carvedilol, metoprolol, timolol | ↑ beta-blockers | Clinical monitoring is recommended for co-administration with beta-blockers that are metabolized by CYP2D6. |
| Calcium channel blockers: e.g. amlodipine, diltiazem, felodipine, nifedipine, verapamil | ↑ calcium channel blockers | Clinical monitoring is recommended for co-administration with calcium channel blockers metabolized by CYP3A. |
| Cardiac Disorders: | ||
| ranolazine, ivabradine | ↑ ranolazine ↑ ivabradine | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions. |
| dronedarone | ↑ dronedarone | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| Other antiarrhythmics | ||
| e.g. amiodarone, disopyramide, flecainide, lidocaine (systemic), mexiletine, propafenone, quinidine | ↑ antiarrhythmics | Clinical monitoring is recommended upon co-administration with antiarrhythmics. |
| digoxin | ↑ digoxin | When co-administering with digoxin, titrate the digoxin dose and monitor digoxin concentrations. |
| Corticosteroids: dexamethasone (systemic) Corticosteroids primarily metabolized by CYP3A: e.g. betamethasone budesonide ciclesonide fluticasone methylprednisolone mometasone triamcinolone | ↓ darunavir ↓ cobicistat ↑ corticosteroids | Co-administration with systemic dexamethasone or other systemic corticosteroids that induce CYP3A may result in loss of therapeutic effect and development of resistance to PREZCOBIX or PREZCOBIX PED. Consider alternative corticosteroids. Co-administration with corticosteroids (all routes of administration) of which exposures are significantly increased by strong CYP3A inhibitors can increase the risk for Cushing's syndrome and adrenal suppression. Alternative corticosteroids including beclomethasone, prednisone and prednisolone (for which PK and/or PD are less affected by strong CYP3A inhibitors relative to other steroids) should be considered, particularly for long-term use. |
| Endothelin receptor antagonists: bosentan | ↓ darunavir ↓ cobicistat ↑ bosentan | Initiation of bosentan in patients taking PREZCOBIX or PREZCOBIX PED : In patients who have been receiving PREZCOBIX or PREZCOBIX PED for at least 10 days, start bosentan at 62.5 mg once daily or every other day based upon individual tolerability. Initiation of PREZCOBIX or PREZCOBIX PED in patients on bosentan : Discontinue use of bosentan at least 36 hours prior to initiation of PREZCOBIX or PREZCOBIX PED. After at least 10 days following the initiation of PREZCOBIX or PREZCOBIX PED, resume bosentan at 62.5 mg once daily or every other day based upon individual tolerability. Switching from darunavir co-administered with ritonavir to PREZCOBIX or PREZCOBIX PED in patients on bosentan : Maintain bosentan dose. |
| Ergot derivatives: e.g. dihydroergotamine, ergotamine, methylergonovine | ↑ ergot derivatives | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues. |
| Hepatitis C virus (HCV): Direct-Acting Antivirals: elbasvir/grazoprevir | ↑ elbasvir/grazoprevir | Co-administration is contraindicated due to potential for the increased risk of alanine transaminase (ALT) elevations. |
| glecaprevir/pibrentasvir | ↑ glecaprevir ↑ pibrentasvir | Co-administration of PREZCOBIX or PREZCOBIX PED with glecaprevir/pibrentasvir is not recommended. |
| Herbal product: St. John's wort ( Hypericum perforatum ) | ↓ darunavir ↓ cobicistat | Co-administration is contraindicated due to potential for reduced plasma concentrations of darunavir, which may result in loss of therapeutic effect and development of resistance. |
| Hormonal contraceptives: | Additional or alternative (non-hormonal) forms of contraception should be considered when estrogen-containing contraceptives are co-administered with PREZCOBIX or PREZCOBIX PED [see Use in Specific Populations (8.3) ] . | |
| drospirenone/ethinylestradiol | ↑ drospirenone ↓ ethinylestradiol | For co-administration with drospirenone, clinical monitoring is recommended due to the potential for hyperkalemia. |
| Other progestin/estrogen contraceptives | progestin: effects unknown estrogen: effects unknown | No data are available to make recommendations on co-administration with other hormonal contraceptives. |
| Immunosuppressants: cyclosporine, sirolimus, tacrolimus | ↑ immunosuppressants | These immunosuppressant agents are metabolized by CYP3A. Therapeutic drug monitoring is recommended with concomitant use |
| Immunosuppressant /neoplastic: everolimus | ↑ immunosuppressants | Co-administration of everolimus and PREZCOBIX or PREZCOBIX PED is not recommended. |
| irinotecan | Discontinue PREZCOBIX or PREZCOBIX PED at least 1 week prior to starting irinotecan therapy. Do not administer PREZCOBIX or PREZCOBIX PED with irinotecan unless there are no therapeutic alternatives. | |
| Inhaled beta agonist: salmeterol | ↑ salmeterol | Co-administration with salmeterol is not recommended and may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations, and sinus tachycardia. |
| Lipid Modifying Agents | ||
| HMG-CoA reductase inhibitors: lovastatin, simvastatin | ↑ lovastatin ↑ simvastatin | Co-administration is contraindicated due to potential for serious reactions such as myopathy including rhabdomyolysis. |
| atorvastatin, fluvastatin, pitavastatin, pravastatin, rosuvastatin | ↑ atorvastatin ↑ fluvastatin ↑ pravastatin ↑ rosuvastatin pitavastatin: effect unknown | For atorvastatin, fluvastatin, pitavastatin, pravastatin, and rosuvastatin, start with the lowest recommended dose and titrate while monitoring for safety (e.g. myopathy). Dosage recommendations with atorvastatin or rosuvastatin are as follows:
|
| Other lipid modifying agents: lomitapide | ↑ lomitapide | Co-administration is contraindicated due to potential for markedly increased transaminases associated with increased plasma concentrations of lomitapide. |
| Narcotic analgesics metabolized by CYP3A: e.g. fentanyl, oxycodone | ↑ fentanyl ↑ oxycodone | Careful monitoring of therapeutic effects and adverse reactions associated with CYP3A-metabolized narcotic analgesics (including potentially fatal respiratory depression) is recommended with co-administration. |
| tramadol | ↑ tramadol | A dose decrease may be needed for tramadol with concomitant use. |
| Narcotic analgesic for treatment of opioid dependence: buprenorphine, buprenorphine/naloxone, methadone | buprenorphine or buprenorphine/ naloxone: effects unknown methadone: effects unknown | Initiation of buprenorphine, buprenorphine/naloxone or methadone in patients taking PREZCOBIX or PREZCOBIX PED : Carefully titrate the dose of buprenorphine, buprenorphine/naloxone or methadone to the desired effect; use the lowest feasible initial or maintenance dose. Initiation of PREZCOBIX or PREZCOBIX PED in patients taking buprenorphine, buprenorphine/naloxone or methadone : A dose adjustment for buprenorphine, buprenorphine/naloxone or methadone may be needed. Monitor clinical signs and symptoms. |
| Opioid Antagonist | ||
| naloxegol | ↑ naloxegol | Co-administration of PREZCOBIX or PREZCOBIX PED and naloxegol is contraindicated due to potential for precipitating opioid withdrawal symptoms. |
| Phosphodiesterase PDE-5 inhibitors: e.g. avanafil, sildenafil, tadalafil, vardenafil | ↑ PDE-5 inhibitors | Co-administration with avanafil is not recommended because a safe and effective avanafil dosage regimen has not been established. Co-administration with PDE-5 inhibitors may result in an increase in PDE-5 inhibitor-associated adverse reactions including hypotension, syncope, visual disturbances and priapism. Use of PDE-5 inhibitors for pulmonary arterial hypertension (PAH): Co-administration with sildenafil used for PAH is contraindicated due to potential for sildenafil associated adverse reactions (which include visual disturbances, hypotension, prolonged erection, and syncope). The following dose adjustments are recommended for use of tadalafil with PREZCOBIX or PREZCOBIX PED:
|
| Platelet aggregation inhibitor: | ||
| ticagrelor | ↑ ticagrelor | Co-administration of PREZCOBIX or PREZCOBIX PED and ticagrelor is not recommended. |
| clopidogrel | ↓ clopidogrel active metabolite | Co-administration of PREZCOBIX or PREZCOBIX PED with clopidogrel is not recommended due to the potential reduction of the antiplatelet activity of clopidogrel. |
| prasugrel | ↔ prasugrel active metabolite | No dose adjustment is needed when prasugrel is co-administered with PREZCOBIX or PREZCOBIX PED. |
| Sedatives/hypnotics: orally administered midazolam, triazolam | ↑ midazolam ↑ triazolam | Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as prolonged or increased sedation or respiratory depression. Triazolam and orally administered midazolam are extensively metabolized by CYP3A. Co-administration of triazolam or orally administered midazolam with PREZCOBIX or PREZCOBIX PED may cause large increases in the concentrations of these benzodiazepines. |
| metabolized by CYP3A: e.g. buspirone, diazepam, estazolam, zolpidem | ↑ sedatives/hypnotics | With concomitant use, titration is recommended with sedatives/hypnotics metabolized by CYP3A and a lower dose of the sedatives/hypnotics should be considered with monitoring for increased and prolonged effects or adverse reactions. |
| parenterally administered midazolam | Co-administration of parenteral midazolam should be done in a setting that ensures close clinical monitoring and appropriate medical management in case of respiratory depression and/or prolonged sedation. Dose reduction for parenteral midazolam should be considered, especially if more than a single dose of midazolam is administered. | |
| Urinary antispasmodics | ||
| fesoterodine | ↑ fesoterodine | When fesoterodine is co-administered with PREZCOBIX or PREZCOBIX PED, do not exceed a fesoterodine dose of 4 mg once daily. |
| solifenacin | ↑ solifenacin | When solifenacin is co-administered with PREZCOBIX or PREZCOBIX PED, do not exceed a solifenacin dose of 5 mg once daily. |
Drugs without Clinically Significant Interactions with PREZCOBIX or PREZCOBIX PED
Clinically relevant drug-drug interactions have not been observed or are not anticipated with concomitant use of darunavir and cobicistat with rilpivirine, dolutegravir, raltegravir, abacavir, emtricitabine, emtricitabine/tenofovir alafenamide, tenofovir DF, lamivudine, stavudine, zidovudine, or acid modifying medications (antacids, H 2 -receptor antagonists, proton pump inhibitors).
DESCRIPTION
PREZCOBIX and PREZCOBIX PED are fixed-dose combination products containing darunavir and cobicistat. Darunavir is an inhibitor of the human immunodeficiency virus (HIV-1) protease. Cobicistat is a mechanism-based inhibitor of cytochrome P450 (CYP) enzymes of the CYP3A family.
Darunavir : Darunavir, in the form of darunavir ethanolate, has the following chemical name: [(1 S ,2 R )-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]-carbamic acid (3 R ,3a S ,6a R )-hexahydrofuro[2,3- b ]furan-3-yl ester monoethanolate. Its molecular formula is C 27 H 37 N 3 O 7 S ∙ C 2 H 5 OH and its molecular weight is 593.73. Darunavir ethanolate has the following structural formula:

Cobicistat : Cobicistat is adsorbed onto silicon dioxide. The chemical name for cobicistat is 1,3-thiazol-5-ylmethyl[(2 R ,5 R )-5-{[(2 S )2-[(methyl{[2-(propan-2-yl)-1,3-thiazol-4-yl]methyl}carbamoyl)amino]-4-(morpholin-4yl)butanoyl]amino}-1,6-diphenylhexan-2-yl]carbamate. It has a molecular formula of C 40 H 53 N 7 O 5 S 2 and a molecular weight of 776.0. It has the following structural formula:

PREZCOBIX ® 800 mg darunavir/150 mg cobicistat tablets are for oral administration. Each tablet contains darunavir ethanolate equivalent to 800 mg of darunavir and 150 mg of cobicistat. The tablets include the following inactive ingredients: colloidal silicon dioxide, crospovidone, hypromellose, magnesium stearate, and silicified microcrystalline cellulose. The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.
PREZCOBIX ® 675 mg darunavir/150 mg cobicistat tablets are for oral administration. Each tablet contains darunavir ethanolate equivalent to 675 mg of darunavir and 150 mg of cobicistat. The tablets include the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. The tablets are film-coated with a coating material containing iron oxide black, iron oxide yellow, polyethylene glycol, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.
PREZCOBIX ® PED 600 mg darunavir/90 mg cobicistat tablets for oral suspension are for oral administration. Each tablet for oral suspension contains darunavir ethanolate equivalent to 600 mg of darunavir and 90 mg of cobicistat. The tablets include the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, silicified microcrystalline cellulose, strawberry flavor, and sucralose. The tablets are film-coated with a coating material containing glycerol monocaprylocaprate type 1, iron oxide red, iron oxide black, macrogol polyvinyl alcohol graft copolymer, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
PREZCOBIX and PREZCOBIX PED are fixed-dose combinations of an HIV-1 antiviral drug, darunavir and a CYP3A inhibitor, cobicistat [see Microbiology (12.4) ].
Pharmacodynamics
Separate thorough QT trials have been conducted for darunavir co-administered with ritonavir and for cobicistat. The effect of darunavir co-administered with cobicistat on the QT interval has not been evaluated.
Darunavir : In a thorough QT/QTc study in 40 healthy participants, darunavir doses (co-administered with 100 mg ritonavir) of approximately 2 times the recommended darunavir dose did not affect the QT/QTc interval.
Cobicistat : The effect of a single dose of cobicistat 250 mg and 400 mg (approximately 1.7 and 2.7 times the recommended dose) on QTc interval was evaluated in a randomized, placebo- and active-controlled (moxifloxacin 400 mg) four-period crossover thorough QT trial in 48 healthy participants. In this trial, no significant QTc prolongation effect of cobicistat was detected. The dose of 400 mg cobicistat is expected to provide information on a high exposure clinical scenario. Prolongation of the PR interval was noted in participants receiving cobicistat in the same trial. The maximum mean (95% upper confidence bound) difference in PR from placebo after baseline-correction was 9.5 (12.1) msec for 250 mg and 20.2 (22.8) msec for 400 mg of cobicistat .
Effects on Serum CreatinineCobicistat : The effect of cobicistat on serum creatinine was investigated in a trial in participants with normal renal function (eGFR ≥80 mL/min, N=12) and mild-to-moderate renal impairment (eGFR 50–79 mL/min, N=18). A statistically significant decrease in the estimated glomerular filtration rate, calculated by Cockcroft-Gault method (eGFR CG ) from baseline, was observed after 7 days of treatment with cobicistat 150 mg among participants with normal renal function (-9.9 ± 13.1 mL/min) and mild-to-moderate renal impairment (-11.9 ± 7.0 mL/min). No statistically significant changes in eGFR CG were observed compared to baseline for participants with normal renal function or mild-to-moderate renal impairment 7 days after cobicistat was discontinued. The actual glomerular filtration rate, as determined by the clearance of probe drug iohexol, was not altered from baseline following treatment of cobicistat among participants with normal renal function and mild-to-moderate renal impairment, indicating that cobicistat inhibits tubular secretion of creatinine, reflected as a reduction in eGFR CG , without affecting the actual glomerular filtration rate.
Pharmacokinetics
The pharmacokinetics of darunavir co-administered with cobicistat (150 mg) have been evaluated in healthy adults and in adults with HIV-1.
Darunavir is primarily metabolized by CYP3A. Cobicistat inhibits CYP3A, thereby increasing the plasma concentrations of darunavir.
Under fed (535 total kcal, 171 kcal from fat, 268 kcal from carbohydrates, 96 kcal from protein) and fasted conditions in healthy participants, the 90% confidence intervals when comparing darunavir exposure between PREZCOBIX (800 mg/150 mg) and darunavir 800 mg co-administered with cobicistat 150 mg as single entities were within 80–125%. No clinically significant difference was observed between PREZCOBIX (675 mg/150 mg) and darunavir 675 mg co-administered with cobicistat 150 mg as single entities under fed conditions in healthy adults. No clinically significant difference was observed between PREZCOBIX PED (600 mg/90 mg) and darunavir suspension (100 mg/mL) at a dose of 600 mg co-administered with cobicistat 90 mg as single entities under fed conditions in healthy adults.
Darunavir exposure when comparing darunavir co-administered with cobicistat (as single entities) to darunavir co-administered with ritonavir was evaluated in a relative bioavailability trial [see cobicistat full prescribing information]. Table 3 displays the pharmacokinetic estimates of darunavir after oral administration of darunavir 800 mg co-administered with ritonavir 100 mg once daily (based on sparse sampling in 335 participants in Trial TMC114-C211 and 280 participants in Trial TMC114-C229) and darunavir 800 mg co-administered with cobicistat 150 mg once daily administered as single entities (based on sparse sampling in 298 participants in Trial GS-US-216-0130) to HIV-1 participants.
| Trial TMC114-C211 (treatment-naïve) Darunavir 800 mg co-administered with ritonavir 100 mg once daily | Trial TMC114-C229 (treatment-experienced) Darunavir 800 mg co-administered with ritonavir 100 mg once daily | Trial GS-US-216-0130 (treatment-naïve and experienced) Darunavir 800 mg co-administered with cobicistat 150 mg once daily | |
|---|---|---|---|
| Parameter | N=335 | N=280 | N=298 |
| AUC 0–24h =area under the concentration-time curve over a 24-hour dosing interval at steady state; C 0h =plasma concentration at the end of a 24-hour dosing interval at steady state; N=number of participants; SD=standard deviation | |||
| AUC 0–24h (ng∙h/mL) | |||
| Mean ± SD | 93026 ± 27050 | 93334 ± 28626 | 100152 ± 32042 |
| Median (Range) | 87854 (45000–219240) | 87788 (45456–236920) | 96900 (34500–224000) |
| C 0h (ng/mL) | |||
| Mean ± SD | 2282 ± 1168 | 2160 ± 1201 | 2043 ± 1257 |
| Median (Range) | 2041 (368–7242) | 1896 (184–7881) | 1875 (70–6890) |
In healthy participants, under fed conditions, when single doses of the darunavir and cobicistat fixed-dose combination tablet were administered, the maximum plasma concentration was achieved within approximately 4 to 4.5 hours for darunavir and approximately 4 to 5 hours for cobicistat.
Effects of Food on Oral AbsorptionWhen compared to fasted conditions, administration of PREZCOBIX (800 mg/150 mg) to healthy adult participants with a high-fat meal (965 total kcal: 129 kcal from protein, 236 kcal from carbohydrates and 600 kcal from fat) resulted in a 70% increase in AUC (0–inf) and a 127% increase in C max for darunavir. Cobicistat exposures were not affected by food. PREZCOBIX or PREZCOBIX PED should be taken with food.
DistributionDarunavir : Darunavir is approximately 95% bound to plasma proteins. Darunavir binds primarily to plasma alpha 1-acid glycoprotein (AAG).
Cobicistat : Cobicistat is 97–98% bound to human plasma proteins and the mean blood–to-plasma ratio was approximately 0.5.
MetabolismDarunavir : In vitro experiments with human liver microsomes (HLMs) indicate that darunavir primarily undergoes oxidative metabolism. Darunavir is extensively metabolized by CYP enzymes, primarily by CYP3A. A mass balance trial in healthy participants showed that after single dose administration of 400 mg 14 C-darunavir co-administered with 100 mg ritonavir, the majority of the radioactivity in the plasma was due to darunavir. At least 3 oxidative metabolites of darunavir have been identified in humans; all showed activity that was at least 90% less than the activity of darunavir against wild-type HIV-1.
Cobicistat : Cobicistat is metabolized by CYP3A and to a minor extent by CYP2D6 enzymes and does not undergo glucuronidation.
EliminationDarunavir : A mass balance trial in healthy participants showed that after single dose administration of 400 mg 14 C-darunavir co-administered with 100 mg ritonavir, approximately 79.5% and 13.9% of the administered dose of 14 C-darunavir was recovered in the feces and urine, respectively. Unchanged darunavir accounted for approximately 41.2% and 7.7% of the administered dose in feces and urine, respectively.
When single doses of the darunavir and cobicistat fixed-dose combination tablet were administered, the terminal elimination half-life of darunavir was approximately 7 hours under fed conditions.
Cobicistat : When single doses of the darunavir and cobicistat fixed-dose combination tablet were administered, the terminal elimination half-life of cobicistat was approximately 4 hours under fed conditions. With single dose administration of 14 C-cobicistat after multiple dosing of cobicistat for six days, the mean percent of the administered dose excreted in feces and urine was 86.2% and 8.2%, respectively.
Specific Populations Hepatic ImpairmentDarunavir : Darunavir is primarily metabolized by the liver. The steady-state pharmacokinetic parameters of darunavir were similar after multiple dose co-administration of darunavir 600 mg co-administered with ritonavir 100 mg twice daily to participants with normal hepatic function (n=16), mild hepatic impairment (Child-Pugh Class A, n=8), and moderate hepatic impairment (Child-Pugh Class B, n=8). The effect of severe hepatic impairment on the pharmacokinetics of darunavir has not been evaluated [see Use in Specific Populations (8.6) ].
Cobicistat : Cobicistat is primarily metabolized by the liver. A trial evaluating the pharmacokinetics of cobicistat was performed in participants with moderate hepatic impairment. No clinically relevant differences in cobicistat pharmacokinetics were observed between participants with moderate hepatic impairment (Child-Pugh Class B) and healthy participants. The effect of severe hepatic impairment on the pharmacokinetics of cobicistat has not been evaluated [see Use in Specific Populations (8.6) ].
Hepatitis B or Hepatitis C Virus Co-InfectionDarunavir : In participants with HIV-1 taking darunavir co-administered with ritonavir, the 48 week analysis of the data from clinical studies in participants with HIV-1 indicated that hepatitis B and/or hepatitis C virus co-infection status had no apparent effect on the exposure of darunavir.
The effect of hepatitis B and/or C virus infection on the pharmacokinetics of PREZCOBIX or PREZCOBIX PED have not been evaluated.
Renal ImpairmentDarunavir : Population pharmacokinetic analysis showed that the pharmacokinetics of darunavir were not significantly affected in participants with HIV-1 with moderate renal impairment taking darunavir co-administered with ritonavir (creatinine clearance between 30–60 mL/min, n=20). There are no pharmacokinetic data available in participants with HIV-1 with severe renal impairment or end stage renal disease taking darunavir co-adminstered with either ritonavir or cobicistat [see Use in Specific Populations (8.7) ].
Cobicistat : A trial of the pharmacokinetics of cobicistat was performed in non-HIV participants with severe renal impairment (estimated creatinine clearance below 30 mL/min). No clinically relevant differences in cobicistat pharmacokinetics were observed between participants with severe renal impairment and healthy participants [see Use in Specific Populations (8.7) ].
GenderDarunavir : In participants with HIV-1 taking darunavir co-administered with ritonavir, population pharmacokinetic analysis showed higher mean darunavir exposure in females compared to males. This difference is not clinically relevant.
Cobicistat : No clinically relevant pharmacokinetic differences have been observed between men and women for cobicistat.
RaceDarunavir : Population pharmacokinetic analysis of darunavir in participants with HIV-1 taking darunavir co-administered with ritonavir indicated that race had no apparent effect on the exposure to darunavir.
Cobicistat : Population pharmacokinetic analysis of cobicistat in participants with HIV-1 indicated that race had no clinically relevant effect on the exposure of cobicistat.
Geriatric PatientsDarunavir : In participants with HIV-1 taking darunavir co-administered with ritonavir, population pharmacokinetic analysis showed no considerable differences in darunavir pharmacokinetics for ages 18 to 75 years compared to ages greater than or equal to 65 years (n=12) [see Use in Specific Populations (8.5) ] .
Cobicistat : Insufficient data are available to determine whether potential differences exist in the pharmacokinetics of cobicistat in geriatric (65 years of age and older) participants compared to younger participants.
Pediatrics Weighing at Least 15 kgAvailable pharmacokinetic data for the different components of PREZCOBIX and PREZCOBIX PED indicate that there were no clinically relevant differences in darunavir exposure between adults and adolescents or children weighing at least 15 kg (see Table 4 ). Cobicistat exposures were similar between adults and adolescents but higher in children aged 3 to less than 12 years, weighing at least 15 kg to <40 kg. The difference observed in children for cobicistat is not considered clinically relevant.
| Parameter | GS-US-216-0128 in Participants Aged ≥3 Years and Weighing ≥ 15 to <25 kg | GS-US-216-0128 in Participants Aged ≥ 6 to < 12 Years and Weighing ≥ 25 to < 40 kg | GS-US-216-0128 in Participants Aged ≥ 12 to < 18 Years and Weighing ≥ 40 kg | TMC114FD2HTX3001 in Adult Participants |
|---|---|---|---|---|
| DRV 600 mg/COBI 90 mg | DRV 675 mg/COBI 150 mg | DRV 800 mg/COBI 150 mg | DRV 800 mg/COBI 150 mg/FTC 200 mg/TAF 10 mg | |
| N=11 | N=8 | N=8 | N=355 | |
| AUC 0–24h =area under the concentration-time curve over a 24-hour dosing interval at steady state; C 0h =plasma concentration at the end of a 24-hour dosing interval at steady state; COBI=cobicistat; DRV=darunavir; FTC=emtricitabine; N=number of participants; TAF=tenofovir alafenamide | ||||
| AUC 0–24h (ng ∙ h/mL) Mean ± standard deviation | 146,477 ± 40,055 | 92,052 ± 17,254 | 69,474 ± 10,359 | 87,909 ± 20,232 |
| C 0h (ng/mL) | 2,265 ± 1,293 | 1,345 ± 569 | 938 ± 445 | 1,899 ± 759 |
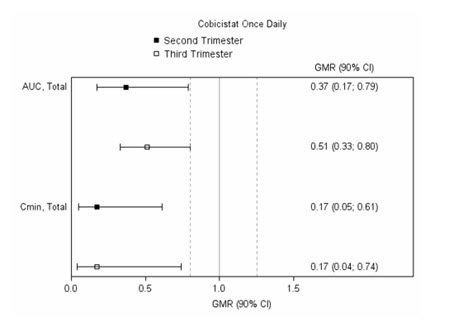
The exposure to total and unbound darunavir boosted with cobicistat after intake of PREZCOBIX (800 mg/150 mg) once daily as part of an antiretroviral regimen was substantially lower during the second and third trimesters of pregnancy compared with 6–12 weeks postpartum (see Table 5 and Figure 1 ).
| Parameter | 2 nd Trimester of pregnancy N=7 | 3 rd Trimester of pregnancy N=6 | Postpartum (6–12 weeks) N=6 |
|---|---|---|---|
| C max =maximum plasma concentration at steady state; AUC 0–24h =area under the concentration-time curve over a 24-hour dosing interval at steady state; C min =minimum plasma concentration at steady state | |||
| C max (ng/mL) Mean ± standard deviation | 4340 ± 1616 | 4910 ± 970 | 7918 ± 2199 |
| AUC 0–24h (ng∙h/mL) | 47293 ± 19058 | 47991 ± 9879 | 99613 ± 34862 |
| C min (ng/mL) | 168 ± 149 | 184 ± 99 | 1538 ± 1344 |
| Legend: 90% CI: 90% confidence interval; GMR: geometric mean ratio (i.e. second or third trimester / postpartum). Solid vertical line: ratio of 1.0; dotted vertical lines: reference lines of 0.8 and 1.25. | |
 |  |
Darunavir is metabolized by CYP3A. Cobicistat is metabolized by CYP3A and, to a minor extent, by CYP2D6. Darunavir co-administered with cobicistat is an inhibitor of CYP3A and CYP2D6. Cobicistat inhibits the following transporters: P-gp, BCRP, MATE1, OATP1B1, and OATP1B3. Based on in vitro data, cobicistat is not expected to induce CYP1A2 or CYP2B6 and based on in vivo data, cobicistat is not expected to induce MDR1 or, in general, CYP3A to a clinically significant extent. The induction effect of cobicistat on CYP2C9, CYP2C19, or UGT1A1 is unknown, but is expected to be low based on CYP3A in vitro induction data [see Drug Interactions (7) ] .
A drug-drug interaction study between darunavir/cobicistat and dabigatran etexilate was conducted in healthy participants. The effects of darunavir on co-administration with dabigatran etexilate are summarized in Table 6.
| Co-administered Drug | Dose/Schedule | N | Effect on Concentration of Co-administered Drug | LS Mean ratio (90% CI) of Co-administered Drug pharmacokinetic parameters with/without darunavir no effect =1.00 | |||
|---|---|---|---|---|---|---|---|
| Co-administered Drug | Darunavir/cobicistat | C max | AUC | C min | |||
| N = number of participants with data q.d. = once daily | |||||||
| Dabigatran etexilate | 150 mg | 800/150 mg single dose | 14 | ↑ | 2.64 (2.29–3.05) | 2.64 (2.32–3.00) | - |
| 800/150 mg q.d. 800/150 mg q.d. for 14 days before co-administered with dabigatran etexilate. | 14 | ↑ | 1.99 (1.72–2.30) | 1.88 (1.65–2.13) | - | ||
Microbiology
Darunavir : Darunavir is an inhibitor of the HIV-1 protease. It selectively inhibits the cleavage of HIV-1 encoded Gag-Pol polyproteins in infected cells, thereby preventing the formation of mature virus particles.
Cobicistat : Cobicistat is a selective, mechanism-based inhibitor of cytochromes P450 of the CYP3A subfamily. Inhibition of CYP3A-mediated metabolism by cobicistat enhances the systemic exposure of CYP3A substrates.
Antiviral ActivityDarunavir : Darunavir exhibits activity against laboratory strains and clinical isolates of HIV-1 and laboratory strains of HIV-2 in acutely infected T-cell lines, human peripheral blood mononuclear cells, and human monocytes/macrophages with median EC 50 values ranging from 1.2 to 8.5 nM (0.7 to 5.0 ng/mL). Darunavir demonstrates antiviral activity in cell culture against a broad panel of HIV-1 group M (A, B, C, D, E, F, G), and group O primary isolates with EC 50 values ranging from less than 0.1 to 4.3 nM. The EC 50 value of darunavir increases by a median factor of 5.4 in the presence of human serum. Darunavir did not show antagonism when studied in combination with the HIV protease inhibitors (PIs) amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, or tipranavir, the N(t)RTIs abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine, or zidovudine, the NNRTIs delavirdine, efavirenz, etravirine, rilpivirine, or nevirapine, and the fusion inhibitor enfuvirtide.
Cobicistat : Cobicistat does not inhibit recombinant HIV-1 protease in a biochemical assay and has no detectable antiviral activity in cell culture against HIV-1. The antiviral activity in cell culture of approved HIV-1 antiretroviral drugs was not antagonized by cobicistat.
Resistance Cell CultureDarunavir : HIV-1 isolates with a decreased susceptibility to darunavir have been selected in cell culture and obtained from participants treated with darunavir co-administered with ritonavir. Darunavir-resistant virus derived in cell culture from wild-type HIV-1 had 21- to 88-fold decreased susceptibility to darunavir and developed 2 to 4 of the following amino acid substitutions S37D, R41E/T, K55Q, H69Q, K70E, T74S, V77I, or I85V in the protease. Selection in cell culture of darunavir resistant HIV-1 from nine HIV-1 strains harboring multiple PI resistance-associated substitutions resulted in the overall emergence of 22 mutations in the protease gene, coding for amino acid substitutions L10F, V11I, I13V, I15V, G16E, L23I, V32I, L33F, S37N, M46I, I47V, I50V, F53L, L63P, A71V, G73S, L76V, V82I, I84V, T91A/S, and Q92R, of which L10F, V32I, L33F, S37N, M46I, I47V, I50V, L63P, A71V, and I84V were the most prevalent. These darunavir-resistant viruses had at least eight protease substitutions and exhibited 50- to 641-fold decreases in darunavir susceptibility with final EC 50 values ranging from 125 nM to 3461 nM.
Clinical StudiesThe resistance profile of PREZCOBIX and PREZCOBIX PED is driven by darunavir. Cobicistat does not select any HIV resistance substitutions, due to its lack of antiviral activity. For the clinical resistance profile of darunavir, refer to the darunavir full prescribing information.
Cross-resistanceCross-resistance among PIs has been observed. Darunavir has a less than 10-fold decreased susceptibility in cell culture against 90% of 3309 clinical isolates resistant to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, and/or tipranavir showing that viruses resistant to these PIs remain susceptible to darunavir. A less than 10-fold decreased susceptibility was observed for the other PIs in 26% to 96% of these PI resistant clinical isolates [nelfinavir (26%), ritonavir (34%), lopinavir (46%), indinavir (57%), atazanavir (59%), saquinavir (64%), amprenavir (70%), and tipranavir (96%)].
Cross-resistance between darunavir and nucleoside/nucleotide reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, fusion inhibitors, CCR5 co-receptor antagonists, or integrase strand transfer inhibitors is unlikely because the viral targets are different.
Baseline Genotype/Phenotype and Virologic Outcome AnalysesBaseline International AIDS Society (IAS)-defined PI resistance substitutions confer reduced virologic response to darunavir. Please refer to the "Baseline Genotype/Phenotype and Virologic Outcome Analyses" section in the darunavir full prescribing information.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Darunavir : Darunavir was evaluated for carcinogenic potential by oral gavage administration to mice and rats up to 104 weeks. Daily doses of 150, 450 and 1000 mg/kg were administered to mice and doses of 50, 150 and 500 mg/kg were administered to rats. A dose-related increase in the incidence of hepatocellular adenomas and carcinomas was observed in males and females of both species and an increase in thyroid follicular cell adenomas was observed in male rats. The observed hepatocellular findings in rodents are considered to be of limited relevance to humans. Repeated administration of darunavir to rats caused hepatic microsomal enzyme induction and increased thyroid hormone elimination, which predispose rats but not humans, to thyroid neoplasms. At the highest tested doses, the systemic exposures to darunavir (based on AUC) were between 0.4- and 0.7-fold (mice) and 0.7- and 1-fold (rats) of exposures observed in humans at the recommended therapeutic doses (darunavir 600 mg co-administered with ritonavir 100 mg twice daily or darunavir 800 mg co-administered with ritonavir 100 mg once daily).
Darunavir was not mutagenic or genotoxic in a battery of in vitro and in vivo assays including bacterial reverse mutation (Ames), chromosomal aberration in human lymphocytes, and in vivo micronucleus test in mice.
Cobicistat : In a long-term carcinogenicity study in mice, no drug-related increases in tumor incidence were observed at doses up to 50 and 100 mg/kg/day in males and females, respectively. Cobicistat exposures at these doses were approximately 7 (male) and 16 (females) times, respectively, the human systemic exposure at the therapeutic daily dose. In a long-term carcinogenicity study of cobicistat in rats, an increased incidence of follicular cell adenomas and/or carcinomas in the thyroid gland was observed at doses of 25 and 50 mg/kg/day in males, and at 30 mg/kg/day in females. The follicular cell findings are considered to be rat-specific, secondary to hepatic microsomal enzyme induction and thyroid hormone imbalance, and are not relevant for humans. At the highest doses tested in the rat carcinogenicity study, systemic exposures were approximately 2 times the human systemic exposure at the therapeutic daily dose.
Cobicistat was not genotoxic in the reverse mutation bacterial test (Ames test), mouse lymphoma or rat micronucleus assays.
Impairment of FertilityDarunavir : No effects on fertility or early embryonic development were observed with darunavir in rats.
Cobicistat : Cobicistat did not affect fertility in male or female rats at daily exposures (AUC) approximately 4-fold higher than human exposures at the recommended 150 mg daily dose.
Fertility was normal in the offspring of rats exposed daily from before birth ( in utero ) through sexual maturity at daily exposures (AUC) of approximately 1.2-fold higher than human exposures at the recommended 150 mg daily dose.
CLINICAL STUDIES
Clinical Trial Results in Adults with HIV-1
The efficacy of PREZCOBIX in adults with HIV-1 is based on efficacy demonstrated in clinical trials of darunavir co-administered with ritonavir [see darunavir full prescribing information].
Clinical Trial Results in Pediatrics with HIV-1
Trial GS-US-216-0128 was a Phase 2/3 multicenter, open-label trial to evaluate the pharmacokinetics, safety, and efficacy of darunavir co-administered with cobicistat in pediatric participants aged 12 years and older (Cohort 1), aged 6 to less than 12 years old (Cohort 2), and aged 3 years and older (Cohort 3), with HIV-1 who were virologically suppressed and had a baseline estimated creatinine clearance ≥90 mL/min/1.73 m 2 . Participants were on a stable antiretroviral regimen including at least 2 NRTIs (for at least 3 months).
Cohort 1: The median age of participants was 14 years (range 12–16 years), median weight was 60 kg (range 45–78 kg), and 43% were male. At baseline, all had plasma HIV-1 RNA <50 copies/mL. At Week 48, 86% (6/7) of participants remained suppressed (HIV-1 RNA <50 copies/mL), and 1 had missing data. From a median baseline CD4+ cell count and CD4+% of 1,117 cells/mm 3 (range 658 to 2,416 cells/mm 3 ) and 45% (range 28% to 56%), respectively, the median change from baseline in CD4+ cell count and CD4+% at Week 48 was -342 cells/mm 3 (range -1,389 to 219 cells/mm 3 ) and -6% (range -12% to 5%), respectively. All 6 participants with available data had CD4+ cell counts above 800 cells/mm 3 at Week 48.
Cohort 2: The median age of participants was 10 years (range 7–11 years), median weight was 31.7 kg (range 29.1–45.4 kg), and 75% were female. At baseline, all had plasma HIV-1 RNA <50 copies/mL. At Week 48, all participants remained suppressed (HIV-1 RNA <50 copies/mL). From a median baseline CD4+ cell count and CD4+% of 998 cells/mm 3 (range 439 to 1113 cells/mm 3 ) and 40% (range 29.1% to 48.5%), respectively, the median change from baseline in CD4+ cell count and CD4+% at Week 48 was -20 cells/mm 3 (range -318 to 245) and 0.75% (-11.40 to 3.20), respectively.
Cohort 3: The median age of participants was 4 years (range: 3 – 7 years), median weight was 17 kg (range 14.7 to 22.0 kg), 55% were female. At baseline, all had plasma HIV-1 RNA <50 copies/mL. At week 48, 91% (10/11) of participants remained suppressed (HIV-1 RNA <50 copies/mL), and 1 had missing data. From a median baseline CD4+ cell count and CD4+% of 1,237 cells/mm 3 (range 705 to 1,636 cells/mm 3 ) and 35.9% (range 25.3% to 49.1%), respectively, the median change from baseline in CD4+ cell count and CD4+% at Week 48 was 34 cells/mm 3 (range -755 to 180) and 1.5% (range -3.00 to 5.50), respectively.
HOW SUPPLIED/STORAGE AND HANDLING
PREZCOBIX ® (darunavir and cobicistat) tablets, 800/150 mg, are supplied as pink, oval-shaped, film-coated tablets debossed with "800" on one side and "TG" on the other side. Bottle of 30 tablets with child-resistant closure (NDC 59676-575-30).
PREZCOBIX ® (darunavir and cobicistat) tablets, 675/150 mg, are supplied as green to dark green, oval-shaped, scored film-coated tablet debossed with "675" on one side and "TG" on the other side. Bottle of 30 tablets with child-resistant closure (NDC 59676-578-30).
Store at 20 °C to 25 °C (between 68 °F to 77 °F); with excursions permitted to 15 °C to 30 °C (59 °F to 86 °F) [see USP Controlled Room Temperature].
PREZCOBIX PED Tablets for Oral SuspensionPREZCOBIX ® PED (darunavir and cobicistat) tablets for oral suspension, 600/90 mg, are supplied as reddish purple, oval-shaped, film-coated tablets debossed with "690" on one side and "TG" on the other side. Bottle of 30 tablets with child-resistant closure (NDC 59676-577-30).
Storage of PREZCOBIX PED tablets for oral suspension:Store in the original container in order to protect from light. Keep the container tightly closed.
Store at 20 °C to 25 °C (between 68 °F to 77 °F); with excursions permitted to 15 °C to 30 °C (59 °F to 86 °F) [see USP Controlled Room Temperature].
Keep PREZCOBIX, PREZCOBIX PED, and all medicines out of reach of children.
| This Instructions for Use has been approved by the U.S. Food and Drug Administration. | Issued: 02/2026 | |
| INSTRUCTIONS FOR USE PREZCOBIX ® PED (prez-koe-bix ped) (darunavir and cobicistat) tablets for oral suspension | ||
This Instructions for Use contains information on how to prepare and give PREZCOBIX PED. Read this Instructions for Use before your child starts taking PREZCOBIX PED for the first time and each time you get a refill. There may be new information. Important information you need to know before giving PREZCOBIX PED:
| ||
Supplies needed to prepare and give PREZCOBIX PED:
| ||
 |
| |
 |
| |
 |
| |
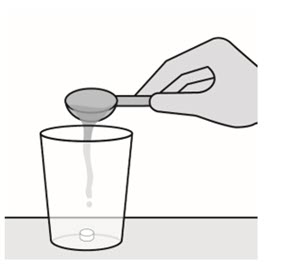 |
| |
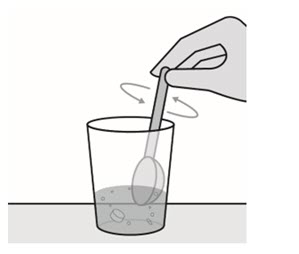 |
| |
| ||
| ||
| ||
Storing PREZCOBIX PED
| ||
| Manufactured for: Janssen Products, LP, Horsham, PA 19044, USA For patent information: www.janssenpatents.com © Johnson & Johnson and its affiliates 2026 For more information call 1-800-526-7736. | ||
Mechanism of Action
PREZCOBIX and PREZCOBIX PED are fixed-dose combinations of an HIV-1 antiviral drug, darunavir and a CYP3A inhibitor, cobicistat [see Microbiology (12.4) ].