Get your patient on Saxenda (Liraglutide)
Saxenda Savings Card, Coupon & Coverage 2026
Saxenda patient education
Patient toolkit
Dosage & administration

Saxenda prescribing information
WARNING: RISK OF THYROID C-CELL TUMORS
- Liraglutide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures in both genders of rats and mice. It is unknown whether SAXENDA causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined [see Warnings and Precautions (5.1 ), Nonclinical Toxicology (13.1 )] .
- SAXENDA is contraindicated in patients with a personal or family history of MTC and in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk of MTC with use of SAXENDA and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with SAXENDA [see Contraindications (4 ), Warnings and Precautions (5.1 )].
INDICATIONS AND USAGE
SAXENDA is indicated in combination with a reduced calorie diet and increased physical activity to reduce excess body weight and maintain weight reduction long term in:
- Adults and pediatric patients aged 12 years and older with body weight greater than 60 kg and obesity.
- Adults with overweight in the presence of at least one weight-related comorbid condition.
Limitations of Use
- SAXENDA contains liraglutide. Coadministration with other liraglutide-containing products or with any other glucagon-like peptide-1 (GLP-1) receptor agonist is not recommended.
- The safety and effectiveness of SAXENDA in pediatric patients with type 2 diabetes have not been established.
DOSAGE AND ADMINISTRATION
- Inject SAXENDA subcutaneously in the abdomen, thigh, or upper arm once daily at any time of day, without regard to the timing of meals. (2.1 )
- The recommended dose of SAXENDA is 3 mg daily. (2.2 )
- Initiate at 0.6 mg per day for one week. In weekly intervals, increase the dose until a dose of 3 mg is reached. (2.2 )
- If pediatric patients do not tolerate an increased dose during dose escalation, the dose may also be lowered to the previous level. Dose escalation for pediatric patients may take up to 8 weeks. (2.2 )
- Pediatric patients who do not tolerate 3 mg daily may have their dose reduced to 2.4 mg daily. (2.2 )
- Adult patients with type 2 diabetes should monitor blood glucose prior to starting SAXENDA and during SAXENDA treatment. (2.2 )
Important Administration Instructions
- Prior to initiation of SAXENDA, train patients on proper injection technique. Refer to the accompanying Instructions for Use for complete administration instructions with illustrations.
- Inspect SAXENDA visually prior to each injection. Only use if solution is clear, colorless, and contains no particles.
- Administer SAXENDA in combination with a reduced-calorie diet and increased physical activity.
- Inject SAXENDA subcutaneously once daily at any time of day, without regard to the timing of meals.
- Inject SAXENDA subcutaneously in the abdomen, thigh, or upper arm. No dosage adjustment is needed if changing the injection site and/or timing.
- Rotate injection sites within the same region in order to reduce the risk of cutaneous amyloidosis [see Adverse Reactions (6.2) ] .
- If a dose is missed, resume the once-daily regimen as prescribed with the next scheduled dose. Do not administer an extra dose or increase the dose to make up for the missed dose.
- If more than 3 days have elapsed since the last SAXENDA dosage, reinitiate SAXENDA at 0.6 mg daily and follow the dosage escalation schedule in Table 1 , to reduce the risk of gastrointestinal adverse reactions associated with reinitiation of treatment.
Dosage in Adults and Pediatric Patients Aged 12 Years and Older
- Initiate SAXENDA with a dose of 0.6 mg daily for one week. Then follow the dosage escalation schedule in Table 1 to reduce the risk of gastrointestinal adverse reactions [see Warnings and Precautions (5.7 ), Adverse Reactions (6.1 )].
Table 1. Dosage Escalation Schedule
Week | Daily Dose |
1 | 0.6 mg |
2 | 1.2 mg |
3 | 1.8 mg |
4 | 2.4 mg |
5 and onward | 3 mg |
- Adult Patients
- For adults, the recommended dosage of SAXENDA is 3 mg daily, lower dosages are for titration only.
- Discontinue SAXENDA if the patient cannot tolerate the 3 mg dosage.
- If patients do not tolerate an increased dose during dosage escalation, consider delaying dosage escalation for approximately one additional week.
- Evaluate the change in body weight 16 weeks after initiating SAXENDA and discontinue SAXENDA if the patient has not lost at least 4% of baseline body weight, since it is unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment.
- In adult patients with type 2 diabetes, monitor blood glucose prior to starting SAXENDA and during SAXENDA treatment.
Pediatric Patients
- For pediatric patients, the recommended maintenance dosage of SAXENDA is 3 mg daily. Pediatric patients who do not tolerate 3 mg daily may have their maintenance dose reduced to 2.4 mg daily. Discontinue SAXENDA if the patient cannot tolerate the 2.4 mg dose.
- If pediatric patients do not tolerate an increased dose during dosage escalation, the dose may also be lowered to the previous level. Dosage escalation for pediatric patients may take up to 8 weeks.
- Evaluate the change in BMI after 12 weeks on the maintenance dose and discontinue SAXENDA if the patient has not had a reduction in BMI of at least 1% from baseline, since it is unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment.
DOSAGE FORMS AND STRENGTHS
Injection: 6 mg/mL clear, colorless solution in a 3 mL prefilled, single-patient-use pen that delivers doses of 0.6 mg, 1.2 mg, 1.8 mg, 2.4 mg, or 3 mg.
USE IN SPECIFIC POPULATIONS
Pregnancy: May cause fetal harm. When pregnancy is recognized, discontinue SAXENDA. (8.1 )
Pregnancy
Risk Summary
Based on animal reproduction studies, there may be risks to the fetus from exposure to SAXENDA during pregnancy. SAXENDA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Additionally, weight loss offers no benefit to a pregnant patient and may cause fetal harm. When a pregnancy is recognized, advise the pregnant patient of the risk to a fetus, and discontinue SAXENDA (see Clinical Considerations ).
Animal reproduction studies identified increased adverse embryofetal developmental outcomes from exposure during pregnancy. Liraglutide exposure was associated with early embryonic deaths and an imbalance in some fetal abnormalities in pregnant rats administered liraglutide during organogenesis at doses that approximate clinical exposures at the maximum recommended human dose (MRHD) of 3 mg/day. In pregnant rabbits administered liraglutide during organogenesis, decreased fetal weight and an increased incidence of major fetal abnormalities were seen at exposures below the human exposures at the MRHD (see Animal Data ).
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage of clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryofetal risk
Appropriate weight gain based on pre-pregnancy weight is currently recommended for all pregnant patients, including those who already have overweight or obesity, because of the obligatory weight gain that occurs in maternal tissues during pregnancy.
Animal Data
Liraglutide has been shown to be teratogenic in rats at or above 0.8-times systemic exposures in obese humans resulting from the maximum recommended human dose (MRHD) of 3 mg/day based on plasma area under the time-concentration curve (AUC) comparison. Liraglutide has been shown to cause reduced growth and increased total major abnormalities in rabbits at systemic exposures below exposure in obese humans at the MRHD based on plasma AUC comparison.
Female rats given subcutaneous doses of 0.1, 0.25 and 1 mg/kg/day liraglutide beginning 2 weeks before mating through gestation day 17 had estimated systemic exposures 0.8-, 3-, and 11-times the exposure in obese humans at the MRHD based on plasma AUC comparison. The number of early embryonic deaths in the 1 mg/kg/day group increased slightly. Fetal abnormalities and variations in kidneys and blood vessels, irregular ossification of the skull, and a more complete state of ossification occurred at all doses. Mottled liver and minimally kinked ribs occurred at the highest dose. The incidence of fetal malformations in liraglutide-treated groups exceeding concurrent and historical controls were misshapen oropharynx and/or narrowed opening into larynx at 0.1 mg/kg/day and umbilical hernia at 0.1 and 0.25 mg/kg/day.
Pregnant rabbits given subcutaneous doses of 0.01, 0.025 and 0.05 mg/kg/day liraglutide from gestation day 6 through day 18 inclusive, had estimated systemic exposures less than the exposure in obese humans at the MRHD of 3 mg/day at all doses, based on plasma AUC comparison. Liraglutide decreased fetal weight and dose-dependently increased the incidence of total major fetal abnormalities at all doses. The incidence of malformations exceeded concurrent and historical controls at 0.01 mg/kg/day (kidneys, scapula), greater than or equal to 0.01 mg/kg/day (eyes, forelimb), 0.025 mg/kg/day (brain, tail and sacral vertebrae, major blood vessels and heart, umbilicus), greater than or equal to 0.025 mg/kg/day (sternum) and at 0.05 mg/kg/day (parietal bones, major blood vessels). Irregular ossification and/or skeletal abnormalities occurred in the skull and jaw, vertebrae and ribs, sternum, pelvis, tail, and scapula; and dose-dependent minor skeletal variations were observed. Visceral abnormalities occurred in blood vessels, lung, liver, and esophagus. Bilobed or bifurcated gallbladder was seen in all treatment groups, but not in the control group.
In pregnant female rats given subcutaneous doses of 0.1, 0.25 and 1 mg/kg/day liraglutide from gestation day 6 through weaning or termination of nursing on lactation day 24, estimated systemic exposures were 0.8-, 3-, and 11-times exposure in obese humans at the MRHD of 3 mg/day, based on plasma AUC comparison. A slight delay in parturition was observed in the majority of treated rats. Group mean body weight of neonatal rats from liraglutide-treated dams was lower than neonatal rats from control group dams. Bloody scabs and agitated behavior occurred in male rats descended from dams treated with 1 mg/kg/day liraglutide. Group mean body weight from birth to postpartum day 14 trended lower in F 2 generation rats descended from liraglutide-treated rats compared to F 2 generation rats descended from controls, but differences did not reach statistical significance for any group.
Lactation
Risk Summary
There are no data on the presence of liraglutide in human milk, the effects on the breastfed infant, or effects on milk production. Liraglutide was present in the milk of lactating rats (see Data ).
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SAXENDA and any potential adverse effects on the breastfed infant from SAXENDA or from the underlying maternal condition.
Data
In lactating rats, liraglutide was present unchanged in milk at concentrations approximately 50% of maternal plasma concentrations.
Pediatric Use
The safety and effectiveness of SAXENDA as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management have been established in pediatric patients aged 12 years and older with body weight above 60 kg and an initial BMI corresponding to 30 kg/m 2 or greater for adults (obese) by international cut-offs. Use of SAXENDA for this indication is supported by a 56-week double-blind, placebo-controlled clinical trial in 251 pediatric patients aged 12 to 17 years, a pharmacokinetic study in pediatric patients, and studies in adults with obesity [see Clinical Pharmacology (12.3 ), Clinical Studies (14.1 , 14.2 )].
In the pediatric clinical trial, one SAXENDA-treated patient had an event of pancreatitis [see Warnings and Precautions (5.2 )] ; more episodes of hypoglycemia confirmed by self blood glucose monitoring occurred in SAXENDA-treated patients compared to placebo [see Warnings and Precautions (5.4 ), Adverse Reactions (6.1 )] ; and mean increases in resting heart rate of 3 to 7 bpm from baseline were observed with SAXENDA-treated patients [see Warnings and Precautions (5.5 )] .
The safety and effectiveness of SAXENDA have not been established in patients less than 12 years of age.
Geriatric Use
In the SAXENDA clinical trials, 232 (6.9%) of the SAXENDA-treated patients were 65 years of age and over, and 17 (0.5%) of the SAXENDA-treated patients were 75 years of age and over. No overall differences in safety or effectiveness were observed between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Renal Impairment
There is limited experience with SAXENDA in patients with mild, moderate, and severe renal impairment, including end‑stage renal disease.
Hepatic Impairment
There is limited experience in patients with mild, moderate, or severe hepatic impairment. Therefore, SAXENDA should be used with caution in this patient population [see Clinical Pharmacology (12.3 )] .
CONTRAINDICATIONS
SAXENDA is contraindicated in:
- Patients with a personal or family history of medullary thyroid carcinoma (MTC) or patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2) [see Warnings and Precautions (5.1 )].
- Patients with a serious hypersensitivity reaction to liraglutide or to any of the excipients in SAXENDA. Serious hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with SAXENDA [see Warnings and Precautions (5.8 )].
WARNINGS AND PRECAUTIONS
- Acute Pancreatitis: Has been observed in patients treated with GLP-1 receptor agonists, including SAXENDA. Discontinue if pancreatitis is suspected. (5.2 )
- Acute Gallbladder Disease: If cholelithiasis or cholecystitis are suspected, gallbladder studies are indicated. (5.3 )
- Hypoglycemia: Can occur in adults when SAXENDA is used with an insulin secretagogue (e.g. a sulfonylurea) or insulin. The risk may be lowered by a reduction in the dose of concomitantly administered insulin secretagogues or insulin. In the pediatric clinical trial, patients did not have type 2 diabetes. Hypoglycemia occurred in SAXENDA-treated pediatric patients. Inform all patients of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia. (5.4 )
- Heart Rate Increase: Monitor heart rate at regular intervals. (5.5 )
- Acute Kidney Injury Due to Volume Depletion: Monitor renal function in patients reporting adverse reactions that could lead to volume depletion. (5.6 )
- Severe Gastrointestinal Adverse Reactions: Use has been associated with gastrointestinal adverse reactions, sometimes severe. SAXENDA is not recommended in patients with severe gastroparesis. (5.7 )
- Hypersensitivity Reactions: Postmarketing reports of serious hypersensitivity reactions (e.g., anaphylactic reactions and angioedema). Discontinue SAXENDA and other suspect medications and promptly seek medical advice. (5.8 )
- Pulmonary Aspiration During General Anesthesia or Deep Sedation: Has been reported in patients receiving GLP-1 receptor agonists undergoing elective surgeries or procedures. Instruct patients to inform healthcare providers of any planned surgeries or procedures. (5.9 )
Risk of Thyroid C-cell Tumors
Liraglutide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors (adenomas and/or carcinomas) at clinically relevant exposures in both genders of rats and mice [see Nonclinical Toxicology (13.1 )] . Malignant thyroid C-cell carcinomas were detected in rats and mice. It is unknown whether SAXENDA will cause thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined.
Cases of MTC in patients treated with liraglutide have been reported in the postmarketing period; the data in these reports are insufficient to establish or exclude a causal relationship between MTC and liraglutide use in humans.
SAXENDA is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2. Counsel patients regarding the potential risk for MTC with the use of SAXENDA and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness).
Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with SAXENDA. Such monitoring may increase the risk of unnecessary procedures, due to low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin may indicate MTC, and patients with MTC usually have calcitonin values greater than 50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
Acute Pancreatitis
Acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with GLP-1 receptor agonists, including liraglutide [see Adverse Reactions (6 )] .
After initiation of SAXENDA, observe patients carefully for signs and symptoms of acute pancreatitis which may include persistent or severe abdominal pain (sometimes radiating to the back) and which may or may not be accompanied by nausea or vomiting. If pancreatitis is suspected, discontinue SAXENDA and initiate appropriate management.
Acute Gallbladder Disease
In SAXENDA clinical trials in adults, 2.2% of SAXENDA-treated patients reported adverse events of cholelithiasis versus 0.8% of placebo-treated patients. The incidence of cholecystitis was 0.8% in SAXENDA-treated patients versus 0.4% in placebo-treated patients. The majority of SAXENDA-treated patients with adverse events of cholelithiasis and cholecystitis required cholecystectomy. Substantial or rapid weight loss can increase the risk of cholelithiasis; however, the incidence of acute gallbladder disease was greater in SAXENDA-treated patients than in placebo-treated patients even after accounting for the degree of weight loss. If cholelithiasis is suspected, gallbladder studies and appropriate clinical follow-up are indicated.
Hypoglycemia
Adult patients with type 2 diabetes mellitus on an insulin secretagogue (e.g., sulfonylurea) or insulin may have an increased risk of hypoglycemia with use of SAXENDA, including severe hypoglycemia. In patients with type 2 diabetes, monitor blood glucose prior to starting SAXENDA and during SAXENDA treatment [see Dosage and Administration (2 ), Adverse Reactions (6.1 )] .
The risk of hypoglycemia may be lowered by a reduction in the dose of sulfonylurea (or other concomitantly administered insulin secretagogues) or insulin. Inform patients using these concomitant medications of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
In the pediatric clinical trial, patients did not have type 2 diabetes but were provided with blood glucose meters. Clinically significant hypoglycemia, defined as blood glucose <54 mg/dL, occurred in 1.6% of the SAXENDA- treated patients compared to 0.8% of placebo-treated patients [see Adverse Reactions (6.1 )] . Inform all pediatric patients of the risk of hypoglycemia and educate them on the signs and symptoms of hypoglycemia.
Heart Rate Increase
Mean increases in resting heart rate of 2 to 3 beats per minute (bpm) were observed with routine clinical monitoring in SAXENDA-treated adult patients compared to placebo in clinical trials. More patients treated with SAXENDA, compared with placebo, had changes from baseline at two consecutive visits of more than 10 bpm (34% versus 19%, respectively) and 20 bpm (5% versus 2%, respectively). At least one resting heart rate exceeding 100 bpm was recorded for 6% of SAXENDA-treated patients compared with 4% of placebo-treated patients, with this occurring at two consecutive study visits for 0.9% and 0.3%, respectively. Tachycardia was reported as an adverse reaction in 0.6% of SAXENDA-treated patients and in 0.1% of placebo-treated patients.
In a clinical pharmacology trial that monitored heart rate continuously for 24 hours, SAXENDA treatment was associated with a heart rate that was 4 to 9 bpm higher than that observed with placebo.
In a pediatric clinical trial, mean increases from baseline in resting heart rate of 3 to 7 bpm were observed with SAXENDA treatment.
Heart rate should be monitored at regular intervals consistent with usual clinical practice. Patients should inform health care providers of palpitations or feelings of a racing heartbeat while at rest during SAXENDA treatment. For patients who experience a sustained increase in resting heart rate while taking SAXENDA, SAXENDA should be discontinued.
Acute Kidney Injury Due to Volume Depletion
There have been postmarketing reports of acute kidney injury, in some cases requiring hemodialysis, in patients treated with liraglutide [see Adverse Reactions (6.2 )] . The majority of the reported events occurred in patients who had experienced gastrointestinal reactions leading to dehydration such as nausea, vomiting, or diarrhea [see Adverse Reactions (6.1 )] . Monitor renal function in patients reporting adverse reactions to SAXENDA that could lead to volume depletion, especially during dosage initiation and escalation [see Use in Specific Populations (8.6 )] .
Severe Gastrointestinal Adverse Reactions
Use of GLP-1 receptor agonists, including liraglutide, has been associated with gastrointestinal adverse reactions, sometimes severe [see Adverse Reactions (6)] . In SAXENDA clinical trials, severe gastrointestinal adverse reactions were reported more frequently among patients receiving SAXENDA (4.8%) than placebo (1.4%). Severe gastrointestinal adverse reactions have also been reported postmarketing with GLP-1 receptor agonists. SAXENDA is not recommended in patients with severe gastroparesis.
Hypersensitivity Reactions
There have been reports of serious hypersensitivity reactions (e.g., anaphylactic reactions and angioedema) in patients treated with SAXENDA [see Contraindications (4 ), Adverse Reactions (6.1 , 6.2 )] . If a hypersensitivity reaction occurs, the patient should discontinue SAXENDA and other suspect medications and promptly seek medical advice.
Anaphylaxis and angioedema have been reported with other GLP-1 receptor agonists. Use caution in a patient with a history of anaphylaxis or angioedema with another GLP-1 receptor agonist because it is unknown whether such patients will be predisposed to these reactions with SAXENDA.
Pulmonary Aspiration During General Anesthesia or Deep Sedation
SAXENDA delays gastric emptying [see Clinical Pharmacology (12.2 )] . There have been rare postmarketing reports of pulmonary aspiration in patients receiving GLP-1 receptor agonists undergoing elective surgeries or procedures requiring general anesthesia or deep sedation who had residual gastric contents despite reported adherence to preoperative fasting recommendations.
Available data are insufficient to inform recommendations to mitigate the risk of pulmonary aspiration during general anesthesia or deep sedation in patients taking SAXENDA, including whether modifying preoperative fasting recommendations or temporarily discontinuing SAXENDA could reduce the incidence of retained gastric contents. Instruct patients to inform healthcare providers prior to any planned surgeries or procedures if they are taking SAXENDA.
ADVERSE REACTIONS
The following serious adverse reactions are described below or elsewhere in the prescribing information:
- Risk of Thyroid C-Cell Tumors [see Warnings and Precautions (5.1 )]
- Acute Pancreatitis [see Warnings and Precautions (5.2 )]
- Acute Gallbladder Disease [see Warnings and Precautions (5.3 )]
- Risk for Hypoglycemia with Concomitant Use of Anti-Diabetic Therapy [see Warnings and Precautions (5.4 )]
- Heart Rate Increase [see Warnings and Precautions (5.5 )]
- Acute Kidney Injury Due to Volume Depletion [see Warnings and Precautions (5.6 )]
- Severe Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.7 )]
- Hypersensitivity Reactions [see Warnings and Precautions (5.8 )]
- Pulmonary Aspiration During General Anesthesia or Deep Sedation [see Warnings and Precautions (5.9 )]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
SAXENDA was evaluated for safety in 5 double-blind, placebo controlled trials that included 3,384 overweight or obese adult patients treated with SAXENDA for a treatment period up to 56 weeks (3 trials), 52 weeks (1 trial), and 32 weeks (1 trial) and one trial of 56 weeks in 125 pediatric patients with obesity aged 12 years and older [see Clinical Studies (14.1 , 14.2 )]. All patients received study drug in addition to a reduced-calorie diet and increased physical activity counseling. In the adult trials, patients received SAXENDA for a mean treatment duration of 46 weeks (median, 56 weeks). Baseline characteristics included a mean age of 47 years, 71% female, 85% white, 39% with hypertension, 15% with type 2 diabetes, 34% with dyslipidemia, 29% with a BMI greater than 40 kg/m 2 , and 9% with cardiovascular disease. In one of the 56-week trials, a subset of patients (with abnormal glucose measurements at randomization) [see Clinical Studies (14.1 )] were enrolled for a placebo-controlled 160-week period instead, followed by a 12-week off-treatment follow-up. For those participating in this 160-week period, patients received SAXENDA for a mean treatment duration of 110 weeks (median, 159 weeks). For all trials, dosing was initiated and increased weekly to reach the 3 mg dose.
In adult clinical trials, 9.8% of patients treated with SAXENDA and 4.3% of patients treated with placebo prematurely discontinued treatment as a result of adverse reactions. The most common adverse reactions leading to discontinuation were nausea (2.9% versus 0.2% for SAXENDA and placebo, respectively), vomiting (1.7% versus less than 0.1%), and diarrhea (1.4% versus 0%).
Adverse reactions reported in greater than or equal to 2% of SAXENDA-treated adult patients and more frequently than in placebo-treated patients are shown in Table 2 . Adverse reactions reported in greater than or equal to 3% of SAXENDA-treated pediatric patients and more frequently than in placebo-treated patients are shown in Table 3 .
- Table 2. Adverse Reactions Occurring in ≥2% of SAXENDA-treated Adult Patients and More Frequently than Placebo
Placebo N=1941 % | SAXENDA N=3384 % | |
| 13.8 | 39.3 |
| 9.9 | 20.9 |
| 8.5 | 19.4 |
| 3.9 | 15.7 |
| 10.5 | 13.9 |
| 12.6 | 13.6 |
| 6.6 | 12.6 |
| 2.7 | 9.6 |
| 4.6 | 7.5 |
| 5 | 6.9 |
| 3.1 | 5.4 |
| 2.2 | 5.3 |
| 2.7 | 5.1 |
| 3.2 | 4.7 |
| 1.7 | 4.7 |
| 3 | 4.5 |
| 0.2 | 4.5 |
| 3.1 | 4.3 |
| 2.5 | 4 |
| 1.6 | 2.8 |
| 1.7 | 2.4 |
| 1 | 2.3 |
| 0.8 | 2.1 |
| 1.6 | 2 |
1 The most common reactions, each reported by 1% to 2.5% of SAXENDA-treated patients and more commonly than by placebo-treated patients, included erythema, pruritus, and rash at the injection site.
2 Defined as blood glucose <54 mg/dL with or without symptoms of hypoglycemia in patients with type 2 diabetes not on concomitant insulin (Study 2, SAXENDA N=423, Placebo N=212). See text below for further information regarding hypoglycemia in patients with and without type 2 diabetes. T2DM = type 2 diabetes mellitus.
Table 3. Adverse Reactions Occurring in ≥3% of SAXENDA-treated Pediatric Patients and More Frequently than Placebo in a 56 Week Clinical Trial
Placebo N=126 % | SAXENDA N=125 % | |
| 14.3 | 42.4 |
| 4 | 34.4 |
| 14.3 | 22.4 |
| 4 | 15.2 |
| 4.8 | 12.8 |
| 3.2 | 10.4 |
| 7.1 | 8 |
| 0.8 | 4.8 |
| 2.4 | 4.8 |
| 3.2 | 4.8 |
| 3.2 | 4.8 |
| 3.2 | 4 |
| 2.4 | 4 |
| 2.4 | 4 |
| 2.4 | 4 |
| 3.2 | 3.2 |
| 0 | 3.2 |
| 2.4 | 3.2 |
| 0.8 | 3.2 |
| 0 | 3.2 |
1 Defined as blood glucose <70 mg/dL with symptoms of hypoglycemia. Pediatric patients did not have type 2 diabetes mellitus. See text below for more detailed hypoglycemia information.
Hypoglycemia
Adult Patients with Type 2 Diabetes
In a clinical trial in adult patients with type 2 diabetes mellitus and overweight (excess weight) or obesity, severe hypoglycemia (defined as requiring the assistance of another person) occurred in 3 (0.7%) of 422 SAXENDA-treated patients (all taking a sulfonylurea) and in none of the 212 placebo-treated patients. In this trial, among patients taking a sulfonylurea, hypoglycemia defined as a plasma glucose less than 54 mg/dL with or without symptoms occurred in 31 (28.2%) of 110 SAXENDA-treated patients and 7 (12.7%) of 55 placebo-treated patients. The doses of sulfonylureas were reduced by 50% at the beginning of the trial per protocol. Among patients not taking a sulfonylurea, blood glucose less than 54 mg/dL with or without symptoms occurred in 22 (7.1%) of 312 SAXENDA-treated patients and 7 (4.5%) of 157 placebo-treated patients.
In a SAXENDA clinical trial in adult patients with overweight (excess weight) or obesity with type 2 diabetes mellitus treated with basal insulin and SAXENDA in combination with a reduced-calorie diet and increased physical activity and up to 2 oral anti-diabetes medications, severe hypoglycemia was reported by 3 (1.5%) of 195 SAXENDA-treated patients and 2 (1%) of 197 placebo-treated patients. No meaningful difference in hypoglycemia, defined as blood glucose less than 54 mg/dL with or without symptoms, was reported between groups.
Adult Patients without Type 2 Diabetes
In SAXENDA clinical trials in adult patients without type 2 diabetes mellitus, there was no systematic capturing or reporting of hypoglycemia; patients were not provided with blood glucose meters or hypoglycemia diaries. Spontaneously reported symptomatic episodes of unconfirmed hypoglycemia were reported by 46 (1.6%) of 2,962 SAXENDA-treated patients and 19 (1.1%) of 1,729 placebo-treated patients. Fasting plasma glucose values obtained at routine clinic visits less than 54 mg/dL, irrespective of hypoglycemic symptoms, occurred in 2 (0.1%) SAXENDA-treated patients and 1 (0.1%) placebo-treated patients.
Pediatric Patients without Type 2 Diabetes
In a 56-week placebo-controlled clinical trial of pediatric patients without type 2 diabetes mellitus in which blood glucose meters were provided, 19 (15.2%) of SAXENDA-treated patients had hypoglycemia with a blood glucose less than 70 mg/dL with symptoms as compared to 5 (4%) of placebo-treated patients. Four (4) events of hypoglycemia defined as a plasma glucose less than 54 mg/dL occurred in 2 (1.6%) of 125 SAXENDA-treated patients and 1 event occurred in 1 (0.8%) of 126 placebo-treated patients. No severe hypoglycemic episodes, defined as requiring assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions, occurred in the SAXENDA treatment group.
Acute Pancreatitis
In SAXENDA clinical trials in adults, acute pancreatitis was confirmed by adjudication in 9 (0.3%) of 3,291 SAXENDA-treated patients and 2 (0.1%) of 1843 placebo-treated patients. In addition, there were 2 cases of acute pancreatitis in SAXENDA-treated patients who prematurely withdrew from these clinical trials, occurring 74 and 124 days after the last dose. There were 2 additional cases in SAXENDA-treated patients, 1 during an off-treatment follow-up period within 2 weeks of discontinuing SAXENDA, and 1 that occurred in a patient who completed treatment and was off-treatment for 106 days.
In a SAXENDA pediatric clinical trial, pancreatitis was not independently adjudicated. Pancreatitis was reported in 1 (0.8%) SAXENDA-treated patient and resulted in treatment discontinuation.
Gastrointestinal Adverse Reactions
In the adult clinical trials, approximately 68% of SAXENDA-treated patients and 39% of placebo-treated patients reported gastrointestinal disorders; the most frequently reported was nausea (39% and 14% of patients treated with SAXENDA and placebo, respectively). Severe gastrointestinal adverse reactions were reported more frequently among patients receiving SAXENDA (4.8%) than placebo (1.4 %). There have been reports of gastrointestinal adverse reactions, such as nausea, vomiting, and diarrhea, associated with volume depletion and renal impairment. Most episodes of gastrointestinal events were mild or moderate and did not lead to discontinuation of therapy (6.2% with SAXENDA versus 0.8% with placebo discontinued treatment as a result of gastrointestinal adverse reactions). The percentage of patients reporting nausea declined as treatment continued. Other common adverse reactions that occurred at a higher incidence among SAXENDA-treated patients included diarrhea, constipation, vomiting, dyspepsia, abdominal pain, dry mouth, gastritis, gastroesophageal reflux disease, flatulence, eructation and abdominal distension. There have been reports of gastrointestinal adverse reactions, such as nausea, vomiting, and diarrhea, associated with volume depletion and renal impairment.
In a pediatric clinical trial, 8% of patients treated with SAXENDA versus no patients who received placebo discontinued treatment as a result of gastrointestinal adverse reactions. Most adverse reactions leading to discontinuation were due to vomiting and nausea (4.8% and 3.2% of SAXENDA-treated patients, respectively).
Cardiovascular Safety
Cardiovascular safety was assessed (LEADER, NCT01179048) in 9,340 patients with inadequately controlled type 2 diabetes and cardiovascular disease randomized to liraglutide 1.8 mg or placebo in addition to standard of care treatments for type 2 diabetes for a median duration of 3.5 years. The primary endpoint was the time from randomization to first occurrence of a major adverse cardiovascular event (MACE) defined as: cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke. No increased risk for MACE was observed with liraglutide 1.8 mg. The total number of primary component MACE endpoints was 1,302 [608 (13.0%) with liraglutide 1.8 mg and 694 (14.9%) with placebo]. Liraglutide 1.8 mg (Victoza) is used in the treatment of type 2 diabetes mellitus in adults. The efficacy of liraglutide at doses below 3 mg daily has not been established for weight reduction.
Asthenia, Fatigue, Malaise, Dysgeusia and Dizziness
Events of asthenia, fatigue, malaise, dysgeusia and dizziness were mainly reported within the first 12 weeks of treatment with SAXENDA and were often co-reported with gastrointestinal events such as nausea, vomiting, and diarrhea.
Immunogenicity
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, the incidence of antibodies to SAXENDA cannot be directly compared with the incidence of antibodies of other products.
Patients treated with SAXENDA may develop anti-liraglutide antibodies. Anti-liraglutide antibodies were detected in 42 (2.8%) of 1505 SAXENDA-treated adult patients with a post-baseline assessment. Antibodies that had a neutralizing effect on liraglutide in an in vitro assay occurred in 18 (1.2%) of 1505 SAXENDA-treated patients. Presence of antibodies may be associated with a higher incidence of injection site reactions and reports of low blood glucose. In clinical trials, these events were usually classified as mild and resolved while patients continued on treatment.
In a pediatric clinical trial, anti-liraglutide antibodies were detected in 14 (12%) of 117 SAXENDA-treated patients with a post-baseline assessment; 5 (4.3%) had persistent antibodies as defined by more than 2 antibody visits at least 16 weeks apart. Two patients (1.7%) remained positive throughout the follow-up period; 1 (0.9%) had antibodies cross reactive to native GLP-1. No patients had neutralizing antibodies.
Allergic Reactions
Urticaria was reported in 0.7% of SAXENDA-treated patients and 0.5% of placebo-treated patients. Anaphylactic reactions, asthma, bronchial hyperreactivity, bronchospasm, oropharyngeal swelling, facial swelling, angioedema, pharyngeal edema, type IV hypersensitivity reactions have been reported in patients treated with liraglutide in clinical trials. Cases of anaphylactic reactions with additional symptoms such as hypotension, palpitations, dyspnea, and edema have been reported with marketed use of liraglutide. Anaphylactic reactions may potentially be life-threatening.
Breast Cancer
In SAXENDA clinical trials in adults, breast cancer confirmed by adjudication was reported in 17 (0.7%) of 2379 SAXENDA-treated women compared with 3 (0.2%) of 1300 placebo-treated women, including invasive cancer (13 SAXENDA- and 2 placebo-treated women) and ductal carcinoma in situ (4 SAXENDA- and 1 placebo-treated woman). The majority of cancers were estrogen- and progesterone-receptor positive. There were too few cases to determine whether these cases were related to SAXENDA. In addition, there are insufficient data to determine whether SAXENDA has an effect on pre-existing breast neoplasia.
Papillary Thyroid Cancer
In SAXENDA clinical trials in adults, papillary thyroid carcinoma confirmed by adjudication was reported in 8 (0.2%) of 3291 SAXENDA-treated patients compared with no cases among 1,843 placebo-treated patients. Four of these papillary thyroid carcinomas were less than 1 cm in greatest diameter and 4 were diagnosed in surgical pathology specimens after thyroidectomy prompted by findings identified prior to treatment.
Colorectal Neoplasms
In SAXENDA clinical trials in adults, benign colorectal neoplasms (mostly colon adenomas) confirmed by adjudication were reported in 20 (0.6%) of 3,291 SAXENDA-treated patients compared with 7 (0.4%) of 1,843 placebo-treated patients. Six positively adjudicated cases of malignant colorectal neoplasms were reported in 5 SAXENDA-treated patients (0.2%, mostly adenocarcinomas) and 1 in a placebo-treated patient (0.1%, neuroendocrine tumor of the rectum).
Cardiac Conduction Disorders
In SAXENDA clinical trials in adults, 11 (0.3%) of 3,384 SAXENDA-treated patients compared with none of the 1,941 placebo-treated patients had a cardiac conduction disorder, reported as first degree atrioventricular block, right bundle branch block, or left bundle branch block.
Hypotension
Adverse reactions related to hypotension (hypotension, orthostatic hypotension, circulatory collapse, and decreased blood pressure) were reported more frequently with SAXENDA (1.1%) compared with placebo (0.5%) in SAXENDA clinical trials in adults. Systolic blood pressure decreases to less than 80 mmHg were observed in 4 (0.1%) SAXENDA-treated patients compared with no placebo-treated patients. One of the SAXENDA-treated patients had hypotension associated with gastrointestinal adverse reactions and renal failure [see Warnings and Precautions (5.6 )] .
Laboratory Abnormalities
Liver Enzymes
Increases in alanine aminotransferase (ALT) greater than or equal to 10 times the upper limit of normal were observed in 5 (0.15%) SAXENDA-treated patients (two of whom had ALT greater than 20 and 40 times the upper limit of normal) compared with 1 (0.05%) placebo-treated patient during the SAXENDA clinical trials. Because clinical evaluation to exclude alternative causes of ALT and aspartate aminotransferase (AST) increases was not done in most cases, the relationship to SAXENDA is uncertain. Some increases in ALT and AST were associated with other confounding factors (such as gallstones).
Serum Calcitonin
Calcitonin, a biological marker of MTC, was measured throughout the clinical development program [see Warnings and Precautions (5.1 )] . More patients treated with SAXENDA in the clinical trials were observed to have high calcitonin values during treatment, compared with placebo. The proportion of patients with calcitonin greater than or equal to 2 times the upper limit of normal at the end of the trial was 1.2% in SAXENDA-treated patients and 0.6% in placebo-treated patients. Calcitonin values greater than 20 ng/L at the end of the trial occurred in 0.5% of SAXENDA-treated patients and 0.2% of placebo-treated patients; among patients with pre-treatment serum calcitonin less than 20 ng/L, none had calcitonin elevations to greater than 50 ng/L at the end of the trial.
Serum Lipase and Amylase
Serum lipase and amylase were routinely measured in the SAXENDA clinical trials. Among SAXENDA-treated patients, 2.1% had a lipase value at anytime during treatment of greater than or equal to 3 times the upper limit of normal compared with 1.0% of placebo-treated patients. 0.1% of SAXENDA-treated patients had an amylase value at anytime in the trial of greater than or equal to 3 times the upper limit of normal versus 0.1% of placebo-treated patients. The clinical significance of elevations in lipase or amylase with SAXENDA is unknown in the absence of other signs and symptoms of pancreatitis [see Warnings and Precautions (5.2 )] .
Postmarketing Experience
The following adverse reactions have been reported during post-approval use of liraglutide, the active ingredient of SAXENDA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal
Acute pancreatitis; hemorrhagic and necrotizing pancreatitis, sometimes resulting in death; ileus, intestinal obstruction, severe constipation including fecal impaction, nausea, vomiting and diarrhea leading to dehydration
Hepatobiliary
- Hyperbilirubinemia, elevations of liver enzymes, cholestasis and hepatitis
Hypersensitivity
- Rash, pruritus, angioedema and anaphylactic reactions
Neoplasms
- Medullary thyroid carcinoma
Neurologic
- Dysesthesia, headache
Pulmonary
- Pulmonary aspiration has occurred in patients receiving GLP-1 receptor agonists undergoing elective surgeries or procedures requiring general anesthesia or deep sedation.
Renal
Acute kidney injury, sometimes requiring hemodialysis; increased serum creatinine
General Disorders and Administration Site Conditions
Allergic reactions: rash and pruritus
Immune System
- Angioedema and anaphylactic reactions
Skin and Subcutaneous Tissue
- Cutaneous amyloidosis, alopecia
DRUG INTERACTIONS
SAXENDA delays gastric emptying. May impact absorption of concomitantly administered oral medications. Use with caution. (7 )
Oral Medications
SAXENDA causes a delay of gastric emptying, and thereby has the potential to impact the absorption of concomitantly administered oral medications. In clinical pharmacology trials, liraglutide did not affect the absorption of the tested orally administered medications to any clinically relevant degree. Nonetheless, monitor for potential consequences of delayed absorption of oral medications concomitantly administered with SAXENDA.
DESCRIPTION
SAXENDA contains liraglutide, an analog of human GLP-1 and acts as a GLP-1 receptor agonist. The peptide precursor of liraglutide, produced by a process that includes expression of recombinant DNA in Saccharomyces cerevisiae, has been engineered to be 97% homologous to native human GLP-1 by substituting arginine for lysine at position 34. Liraglutide is made by attaching a C-16 fatty acid (palmitic acid) with a glutamic acid spacer on the remaining lysine residue at position 26 of the peptide precursor. The molecular formula of liraglutide is C 172 H 265 N 43 O 51 and the molecular weight is 3751.2 Daltons. The structural formula ( Figure 1 ) is:
Figure 1. Structural Formula of liraglutide
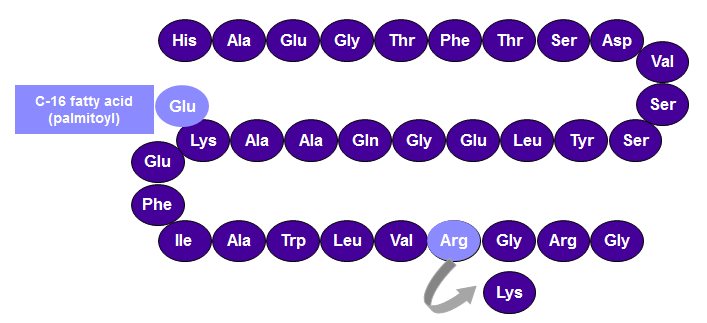
SAXENDA injection is a sterile, aqueous, clear, colorless or almost colorless solution for subcutaneous use. Each 1 mL of SAXENDA solution contains 6 mg of liraglutide and the following inactive ingredients: disodium phosphate dihydrate, 1.42 mg; propylene glycol, 14 mg; phenol, 5.5 mg; and water for injection. SAXENDA has a pH of approximately 8.15, hydrochloric acid or sodium hydroxide may be added to adjust pH. Each prefilled pen contains a 3 mL solution of SAXENDA equivalent to 18 mg liraglutide (free-base, anhydrous).
CLINICAL PHARMACOLOGY
Mechanism of Action
Liraglutide is an acylated human GLP-1 receptor agonist with 97% amino acid sequence homology to endogenous human GLP-1(7-37). Like endogenous GLP-1, liraglutide binds to and activates the GLP-1 receptor, a cell-surface receptor coupled to adenylyl cyclase activation through the stimulatory G-protein, Gs. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the ubiquitous endogenous enzymes, dipeptidyl peptidase 4 (DPP-4) and neutral endopeptidases (NEP). Unlike native GLP-1, liraglutide is stable against metabolic degradation by both peptidases and has a plasma half-life of 13 hours after subcutaneous administration. The pharmacokinetic profile of liraglutide, which makes it suitable for once-daily administration, is a result of self-association that delays absorption, plasma protein binding, and stability against metabolic degradation by DPP-4 and NEP.
GLP-1 is a physiological regulator of appetite and calorie intake, and the GLP-1 receptor is present in several areas of the brain involved in appetite regulation. In animal studies, peripheral administration of liraglutide resulted in the presence of liraglutide in specific brain regions regulating appetite, including the hypothalamus. Although liraglutide activated neurons in brain regions known to regulate appetite, specific brain regions mediating the effects of liraglutide on appetite were not identified in rats.
Pharmacodynamics
Liraglutide lowers body weight through decreased calorie intake. Liraglutide does not increase 24-hour energy expenditure.
As with other GLP-1 receptor agonists, liraglutide stimulates insulin secretion and reduces glucagon secretion in a glucose-dependent manner. These effects can lead to a reduction of blood glucose.
Gastric Emptying
Liraglutide delays gastric emptying.
Cardiac Electrophysiology (QTc) in healthy volunteers
The effect of liraglutide on cardiac repolarization was tested in a QTc study. Liraglutide at steady-state concentrations after daily doses up to 1.8 mg did not produce QTc prolongation. The maximum liraglutide plasma concentration (C max ) in overweight and obese subjects treated with liraglutide 3 mg is similar to the C max observed in the liraglutide QTc study in healthy volunteers.
Pharmacokinetics
Absorption - Following subcutaneous administration, maximum concentrations of liraglutide are achieved at 11 hours post dosing. The average liraglutide steady state concentration (AUCτ /24 ) reached approximately 116 ng/mL in obese (BMI 30-40 kg/m 2 ) subjects following administration of SAXENDA. Liraglutide exposure increased proportionally in the dose range of 0.6 mg to 3 mg. The intra-subject coefficient of variation for liraglutide AUC was 11% following single dose administration. Liraglutide exposures were considered similar among three subcutaneous injection sites (upper arm, abdomen, and thigh). Absolute bioavailability of liraglutide following subcutaneous administration is approximately 55%.
Distribution - The mean apparent volume of distribution after subcutaneous administration of liraglutide 3 mg is 20 to 25L (for a person weighing approximately 100 kg). The mean volume of distribution after intravenous administration of liraglutide is 0.07 L/kg. Liraglutide is extensively bound to plasma protein (greater than 98%).
Metabolism - During the initial 24 hours following administration of a single [3H]-liraglutide dose to healthy subjects, the major component in plasma was intact liraglutide. Liraglutide is endogenously metabolized in a similar manner to large proteins without a specific organ as a major route of elimination.
Elimination - Following a [3H]-liraglutide dose, intact liraglutide was not detected in urine or feces. Only a minor part of the administered radioactivity was excreted as liraglutide-related metabolites in urine or feces (6% and 5%, respectively). The majority of urine and feces radioactivity was excreted during the first 6 to 8 days. The mean apparent clearance following subcutaneous administration of a single dose of liraglutide is approximately 0.9 to 1.4 L/h with an elimination half-life of approximately 13 hours, making liraglutide suitable for once daily administration.
Specific Populations
Elderly - Age had no effect on the pharmacokinetics of liraglutide based on a pharmacokinetic study in healthy elderly subjects (65 to 83 years) and population pharmacokinetic analyses of data from overweight and obese patients 18 to 82 years of age [see Use in Specific Populations (8.5 )] .
Gender - Based on the results of population pharmacokinetic analyses, females have 24% lower weight adjusted clearance of SAXENDA compared to males.
Race and Ethnicity - Race and ethnicity had no effect on the pharmacokinetics of liraglutide based on the results of population pharmacokinetic analyses that included overweight and obese patients of White, Black or African American, Asian and Hispanic/Non-Hispanic Ethnic groups.
Body Weight - Body weight significantly affects the pharmacokinetics of liraglutide based on results of population pharmacokinetic analyses conducted in patients with body weight range of 60 to 234 kg. The exposure of liraglutide decreases as baseline body weight increases.
Pediatric - A population pharmacokinetic analysis was conducted for SAXENDA using data from 134 pediatric patients (12 to 17 years of age) with obesity. The liraglutide exposure in the pediatric patients was similar to that in adults with obesity [see Use in Specific Populations (8.4 )] .
Renal Impairment - The single-dose pharmacokinetics of liraglutide were evaluated in patients with varying degrees of renal impairment. Patients with mild (estimated creatinine clearance 50-80 mL/min) to severe (estimated creatinine clearance less than 30 mL/min) renal impairment and patients with end-stage renal disease requiring dialysis were included in the trial. Compared to healthy subjects, liraglutide AUC in mild, moderate, and severe renal impairment and in end-stage renal disease was on average 35%, 19%, 29% and 30% lower, respectively [see Use in Specific Populations (8.6 )] .
Hepatic Impairment - The single-dose pharmacokinetics of liraglutide were evaluated in patients with varying degrees of hepatic impairment. Patients with mild (Child Pugh score 5 to 6) to severe (Child Pugh score greater than 9) hepatic impairment were included in the trial. Compared to healthy subjects, liraglutide AUC in subjects with mild, moderate and severe hepatic impairment was on average 11%, 14% and 42% lower, respectively [see Use in Specific Populations (8.7)].
Drug Interactions
In vitro assessment of drug-drug interactions
Liraglutide has low potential for pharmacokinetic drug-drug interactions related to cytochrome P450 (CYP) and plasma protein binding.
In vivo assessment of drug-drug interactions
The drug-drug interaction studies were performed at steady state with liraglutide 1.8 mg/day. The effect on rate of gastric emptying was equivalent between liraglutide 1.8 mg and 3 mg (acetaminophen AUC 0-300min ).
Administration of the interacting drugs was timed so that C max of liraglutide (8-12 h) would coincide with the absorption peak of the co-administered drugs.
Oral Contraceptives
A single dose of an oral contraceptive combination product containing 0.03 mg ethinylestradiol and 0.15 mg levonorgestrel was administered under fed conditions and 7 hours after the dose of liraglutide at steady state. Liraglutide lowered ethinylestradiol and levonorgestrel C max by 12% and 13%, respectively. There was no effect of liraglutide on the overall exposure (AUC) of ethinylestradiol. Liraglutide increased the levonorgestrel AUC 0-∞ by 18%. Liraglutide delayed T max for both ethinylestradiol and levonorgestrel by 1.5 h.
Digoxin
A single dose of digoxin 1 mg was administered 7 hours after the dose of liraglutide at steady state. The concomitant administration with liraglutide resulted in a reduction of digoxin AUC by 16%; C max decreased by 31%. Digoxin median time to maximal concentration (T max ) was delayed from 1 h to 1.5 h.
Lisinopril
A single dose of lisinopril 20 mg was administered 5 minutes after the dose of liraglutide at steady state. The coadministration with liraglutide resulted in a reduction of lisinopril AUC by 15%; C max decreased by 27%. Lisinopril median T max was delayed from 6 h to 8 h with liraglutide.
Atorvastatin
Liraglutide did not change the overall exposure (AUC) of atorvastatin following a single dose of atorvastatin 40 mg, administered 5 hours after the dose of liraglutide at steady state. Atorvastatin C max was decreased by 38% and median T max was delayed from 1 h to 3 h with liraglutide.
Acetaminophen
Liraglutide did not change the overall exposure (AUC) of acetaminophen following a single dose of acetaminophen 1,000 mg, administered 8 hours after the dose of liraglutide at steady state. Acetaminophen C max was decreased by 31% and median T max was delayed up to 15 minutes.
Griseofulvin
Liraglutide did not change the overall exposure (AUC) of griseofulvin following co-administration of a single dose of griseofulvin 500 mg with liraglutide at steady state. Griseofulvin C max increased by 37% while median T max did not change.
Insulin Detemir
No pharmacokinetic interaction was observed between liraglutide and insulin detemir when separate subcutaneous injections of insulin detemir 0.5 Unit/kg (single-dose) and liraglutide 1.8 mg (steady state) were administered to patients with type 2 diabetes mellitus.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
A 104-week carcinogenicity study was conducted in male and female CD-1 mice at doses of 0.03, 0.2, 1, and 3 mg/kg/day liraglutide administered by bolus subcutaneous injection yielding systemic exposures 0.2-, 2-, 10- and 43-times the exposure in obese humans, respectively, at the maximum recommended human dose (MRHD) of 3 mg/day based on plasma AUC comparison. A dose-related increase in benign thyroid C-cell adenomas was seen in the 1 and the 3 mg/kg/day groups with incidences of 13% and 19% in males and 6% and 20% in females, respectively. C-cell adenomas did not occur in control groups or 0.03 and 0.2 mg/kg/day groups. Treatment-related malignant C-cell carcinomas occurred in 3% of females in the 3 mg/kg/day group. Thyroid C-cell tumors are rare findings during carcinogenicity testing in mice. A treatment-related increase in fibrosarcomas was seen on the dorsal skin and subcutis, the body surface used for drug injection, in males in the 3 mg/kg/day group. These fibrosarcomas were attributed to the high local concentration of drug near the injection site. The liraglutide concentration in the clinical formulation (6 mg/mL) is 10-times higher than the concentration in the formulation used to administer 3 mg/kg/day liraglutide to mice in the carcinogenicity study (0.6 mg/mL).
A 104-week carcinogenicity study was conducted in male and female Sprague Dawley rats at doses of 0.075, 0.25 and 0.75 mg/kg/day liraglutide administered by bolus subcutaneous injection with exposures 0.5-, 2- and 7-times the exposure in obese humans, respectively, resulting from the MRHD based on plasma AUC comparison. A treatment-related increase in benign thyroid C-cell adenomas was seen in males in 0.25 and 0.75 mg/kg/day liraglutide groups with incidences of 12%, 16%, 42%, and 46% and in all female liraglutide-treated groups with incidences of 10%, 27%, 33%, and 56% in 0 (control), 0.075, 0.25, and 0.75 mg/kg/day groups, respectively. A treatment-related increase in malignant thyroid C-cell carcinomas was observed in all male liraglutide-treated groups with incidences of 2%, 8%, 6%, and 14% and in females at 0.25 and 0.75 mg/kg/day with incidences of 0%, 0%, 4%, and 6% in 0 (control), 0.075, 0.25, and 0.75 mg/kg/day groups, respectively. Thyroid C-cell carcinomas are rare findings during carcinogenicity testing in rats.
Studies in mice demonstrated that liraglutide-induced C-cell proliferation was dependent on the GLP-1 receptor and that liraglutide did not cause activation of the REarranged during Transfection (RET) proto-oncogene in thyroid C-cells.
Human relevance of thyroid C-cell tumors in mice and rats is unknown and has not been determined by clinical studies or nonclinical studies [see Boxed Warning, Warnings and Precautions (5.1 )] .
Liraglutide was negative with and without metabolic activation in the Ames test for mutagenicity and in a human peripheral blood lymphocyte chromosome aberration test for clastogenicity. Liraglutide was negative in repeat-dose in vivo micronucleus tests in rats.
In rat fertility studies using subcutaneous doses of 0.1, 0.25 and 1 mg/kg/day liraglutide, males were treated for 4 weeks prior to and throughout mating and females were treated 2 weeks prior to and throughout mating until gestation day 17. No direct adverse effects on male fertility was observed at doses up to 1 mg/kg/day, a high dose yielding an estimated systemic exposure 11-times the exposure in obese humans at the MRHD, based on plasma AUC comparison. In female rats, an increase in early embryonic deaths occurred at 1 mg/kg/day. Reduced body weight gain and food consumption were observed in females at the 1 mg/kg/day dose.
CLINICAL STUDIES
Weight Management Trials in Adults with Overweight or Obesity
The safety and efficacy of SAXENDA for chronic weight management in conjunction with reduced caloric intake and increased physical activity were studied in three 56-week, randomized, double-blind, placebo-controlled trials. In all studies, SAXENDA was titrated to 3 mg daily during a 4-week period. All patients received instruction for a reduced-calorie diet (approximately 500 kcal/day deficit) and physical activity counseling (recommended increase in physical activity of minimum 150 mins/week) that began with the first dose of study medication or placebo and continued throughout the trial.
Study 1 enrolled 3,731 patients with obesity (BMI greater than or equal to 30 kg/m 2 ) or with overweight (BMI 27-29.9 kg/m 2 ) and at least one weight-related comorbid condition such as treated or untreated dyslipidemia or hypertension; patients with type 2 diabetes mellitus were excluded. Patients were randomized in a 2:1 ratio to either SAXENDA or placebo. Patients were stratified based on the presence or absence of abnormal blood glucose measurements at randomization. All patients were treated for up to 56 weeks. Those patients with abnormal glucose measurements at randomization (2,254 of the 3731 patients) were treated for a total of 160 weeks. At baseline, mean age was 45 years (range 18 to 78), 79% were female, 85% were White, 10% were Black or African American, and 11% were Hispanic/Latino Ethnicity. Mean baseline body weight was 106.3 kg, and mean BMI was 38.3 kg/m 2 .
Study 2 was a 56-week trial that enrolled 635 patients with type 2 diabetes and with either overweight or obesity (as defined above). Patients were to have an HbA 1c of 7% to 10% and be treated with metformin, a sulfonylurea, or a glitazone as single agent or in any combination, or with a reduced-calorie diet and physical activity alone. Patients were randomized in a 2:1 ratio to receive either SAXENDA or placebo. The mean age was 55 years (range 18 to 82), 50% were female, 83% were White, 12% were Black or African American, and 10% were Hispanic/Latino Ethnicity. Mean baseline body weight was 105.9 kg and mean BMI was 37.1 kg/m 2 .
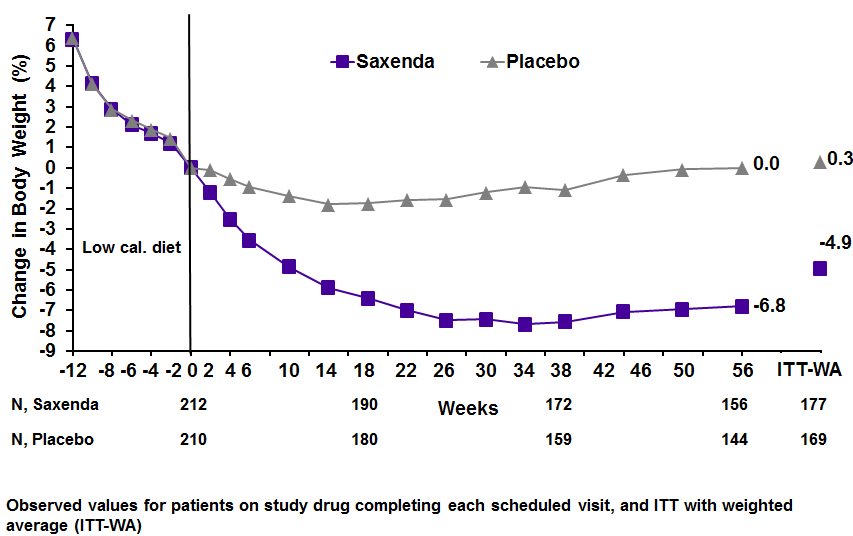
Study 3 was a 56-week trial that enrolled 422 patients with obesity (BMI greater than or equal to 30 kg/m 2 ) or with overweight (BMI 27-29.9 kg/m 2 ) and at least one weight-related comorbid condition such as treated or untreated dyslipidemia or hypertension; patients with type 2 diabetes mellitus were excluded. All patients were first treated with a low-calorie diet (total energy intake 1,200-1,400 kcal/day) in a run-in period lasting up to 12 weeks. Patients who lost at least 5% of their screening body weight after 4 to 12 weeks during the run-in were then randomized, with equal allocation, to receive either SAXENDA or placebo for 56 weeks. The mean age was 46 years (range 18 to 73), 81% were female, 84% were White, 13% were Black or African American, and 7% were Hispanic/Latino Ethnicity. Mean baseline body weight was 99.6 kg and mean BMI was 35.6 kg/m 2 .
The proportions of patients who discontinued study drug in the 56-week trials were 27% for the SAXENDA-treated group and 35% for the placebo-treated group, and in the 160-week trial the proportions of patients who discontinued were 47% and 55%, respectively. In the 56-week trials, approximately 10% of patients treated with SAXENDA and 4% of patients treated with placebo discontinued treatment due to an adverse reaction [see Adverse Reactions (6.1 )] . The majority of patients who discontinued SAXENDA due to adverse reactions did so during the first few months of treatment. In the 160-week trial the proportions of patients who discontinued due to an adverse reaction was 13% and 6% for SAXENDA- and placebo-treated patients, respectively.
Effect of SAXENDA on Body Weight in 56-week Trials
For Study 1 and Study 2, the primary efficacy parameters were mean percent change in body weight and the percentages of patients achieving greater than or equal to 5% and 10% weight loss from baseline to Week 56. For Study 3, the primary efficacy parameters were mean percent change in body weight from randomization to Week 56, the percentage of patients not gaining more than 0.5% body weight from randomization (i.e., after run-in) to Week 56, and the percentage of patients achieving greater than or equal to 5% weight loss from randomization to Week 56. Because losing at least 5% of fasting body weight through lifestyle intervention during the 4- to 12-week run-in was a condition for their continued participation in the randomized treatment period, the results may not reflect those expected in the general population.
Table 4 presents the results for the changes in weight observed in Studies 1, 2, and 3. After 56 weeks, treatment with SAXENDA resulted in a statistically significant reduction in weight compared with placebo. Statistically significantly greater proportions of patients treated with SAXENDA achieved 5% and 10% weight loss than those treated with placebo. In Study 3, statistically significantly more patients randomized to SAXENDA than placebo had not gained more than 0.5% of body weight from randomization to Week 56.
Table 4. Changes in Weight at Week 56 for Studies 1, 2, and 3
Study 1 (Obesity or overweight with comorbidity) | Study 2 (Type 2 diabetes with obesity or overweight) | Study 3 (Obesity or overweight with comorbidity following at least 5% weight loss with diet) | ||||
SAXENDA N=2487 | Placebo N=1244 | SAXENDA N=423 | Placebo N=212 | SAXENDA N=212 | Placebo N=210 | |
Weight | ||||||
| 106.2 (21.2) | 106.2 (21.7) | 105.7 (21.9) | 106.5 (21.3) | 100.4 (20.8) | 98.7 (21.2) |
| -7.4 | -3 | -5.4 | -1.7 | -4.9 | 0.3 |
| -4.5 • (-5.2; -3.8) | -3.7 • (-4.7; -2.7) | -5.2 • (-6.8; -3.5) | |||
% of Patients losing greater than or equal to 5% body weight | 62.3% | 34.4% | 49% | 16.4% | 44.2% | 21.7% |
| 27.9 • (23.9; 31.9) | 32.6 • (25.1; 40.1) | 22.6 • (13.9; 31.3) | |||
% of Patients losing greater than 10% body weight | 33.9% | 15.4% | 22.4% | 5.5% | 25.4% | 6.9% |
| 18.5 • (15.2; 21.7) | 16.9 • (11.7; 22.1) | 18.5 • (11.7; 25.3) | |||
SD = Standard Deviation; CI = Confidence Interval
• p < 0.0001 compared to placebo. Type 1 error was controlled across the three endpoints.
Includes all randomized subjects who had a baseline body weight measurement. All available body weight data during the 56 week treatment period are included in the analysis. In Studies 1 and 2 missing values for Week 56 were handled using multiple imputations analysis. In Study 3 missing values for Week 56 were handled using weighted regression analysis.
The cumulative frequency distributions of change in body weight from baseline to Week 56 are shown in Figure 2 for Studies 1 and 2. One way to interpret this figure is to select a change in body weight of interest on the horizontal axis and note the corresponding proportions of patients (vertical axis) in each treatment group who achieved at least that degree of weight loss. For example, note that the vertical line arising from ‑10% in Study 1 intersects the SAXENDA and placebo curves at approximately 34% and 15%, respectively, which correspond to the values shown in Table 4 .
Figure 2. Change in body weight (%) from baseline to Week 56 (Study 1 on left and Study 2 on right) 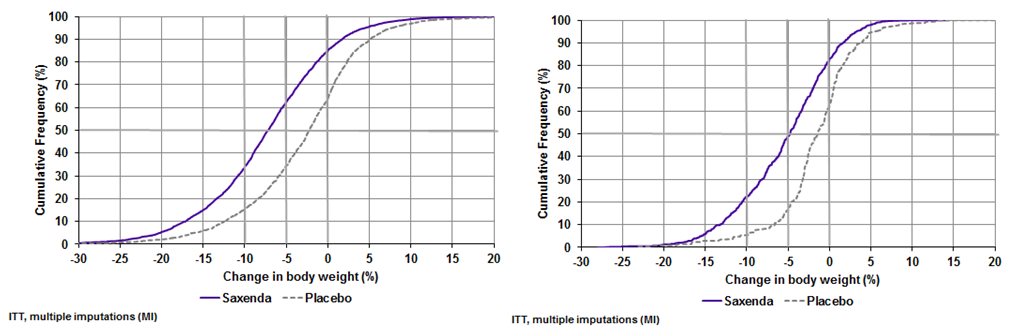
The time courses of weight loss with SAXENDA and placebo from baseline through Week 56 are depicted in Figure 3 and Figure 4 .
Figure 3. Change from baseline (%) in body weight (Study 1 on left and Study 2 on right)
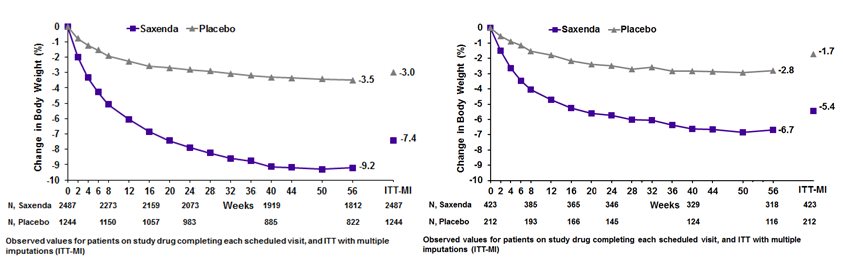
Figure 4. Change from baseline (%) in body weight during Study 3
Effect of SAXENDA on Body Weight in a 160-week Trial (Study 1, Subset of Patients with Abnormal Blood Glucose at Randomization)
The numbers and percentages of patients known to have lost greater than or equal to 5% body weight at Week 56 and/or Week 160 in Study 1 (patients with abnormal glucose at randomization only) are summarized in Table 5 for descriptive purposes.
- Table 5. Changes in Weight at Week 56 and Week 160 for Study 1 (Subset of Patients with Abnormal Blood Glucose at Randomization)
SAXENDA N=1505 | Placebo N=749 | |
Baseline mean body weight (SD) (kg) | 107.5 (21.6) | 107.9 (21.8) |
Number (%) of patients known to lose greater than or equal to 5% body weight at 56 weeks | 817 (56%) | 182 (25%) |
Number (%) of patients known to lose greater than or equal to 5% body weight at 160 weeks | 424 (28%) | 102 (14%) |
Number (%) of patients known to lose greater than or equal to 5% body weight at both 56 weeks and 160 weeks | 391 (26%) | 74 (10%) |
Number (%) of patients with weight assessment at 160 weeks | 747 (50%) | 322 (43%) |
SD = Standard Deviation
Includes all randomized subjects who had a baseline body weight measurement. All available body weight data at 56 and 160 weeks are included in the analysis.
Effect of SAXENDA on Anthropometry and Cardiometabolic Parameters in 56-week Trials
Changes in waist circumference and cardiometabolic parameters with SAXENDA are shown in Table 6 for Study 1 (patients without diabetes mellitus) and Table 7 for Study 2 (patients with type 2 diabetes). Results from Study 3, which also enrolled patients without diabetes mellitus, were similar to Study 1.
- Table 6. Mean Changes in Anthropometry and Cardiometabolic Parameters in Study 1 (Patients without Diabetes)
SAXENDA N=2487 | Placebo N=1244 | ||||
Baseline | Change from Baseline (LSMean 1 ) | Baseline | Change from Baseline (LSMean 1 ) | SAXENDA minus Placebo (LSMean) | |
Waist Circumference (cm) | 115 | -8.2 | 114.5 | -4 | -4.2 |
Systolic Blood Pressure (mmHg) | 123 | -4.3 | 123.3 | -1.5 | -2.8 |
Diastolic Blood Pressure (mmHg) | 78.7 | -2.7 | 78.9 | -1.8 | -0.9 |
Heart Rate (bpm) 2 | 71.4 | 2.6 | 71.3 | 0.1 | 2.5 |
HbA 1c (%) | 5.6 | -0.3 | 5.6 | -0.1 | -0.2 |
Baseline | % Change from Baseline (LSMean 1 ) | Baseline | % Change from Baseline (LSMean 1 ) | Relative Difference of SAXENDA to Placebo (LSMean) | |
Total Cholesterol (mg/dL) • | 193.8 | -3.2 | 194.4 | -0.9 | -2.3 |
LDL Cholesterol (mg/dL) • | 111.8 | -3.1 | 112.3 | -0.7 | -2.4 |
HDL Cholesterol (mg/dL) • | 51.4 | 2.3 | 50.9 | 0.5 | 1.9 |
Triglycerides (mg/dL) † | 125.7 | -13 | 128.3 | -4.1 | -7.1 |
- Based on last observation carried forward method while on study drug
- 1 Least squares mean adjusted for treatment, country, sex, pre-diabetes status at screening, baseline BMI stratum and an interaction between pre-diabetes status at screening and BMI stratum as fixed factors, and the baseline value as covariate.
- 2 See Warnings and Precautions (5.5 )
- • Baseline value is the geometric mean
- † Values are baseline median, median % change, and the Hodges-Lehmann estimate of the median treatment difference.
Table 7. Mean Changes in Anthropometry and Cardiometabolic Parameters in Study 2 (Patients with Diabetes Mellitus)
SAXENDA N=423 | Placebo N=212 | ||||
Baseline | Change from Baseline (LSMean 1 ) | Baseline | Change From Baseline (LSMean 1 ) | SAXENDA minus Placebo (LSMean) | |
Waist Circumference (cm) | 118.1 | -6 | 117.3 | -2.8 | -3.2 |
Systolic Blood Pressure (mmHg) | 128.9 | -3 | 129.2 | -0.4 | -2.6 |
Diastolic Blood Pressure (mmHg) | 79 | -1 | 79.3 | -0.6 | -0.4 |
Heart Rate (bpm) 2 | 74 | 2 | 74 | -1.5 | 3.4 |
HbA 1c (%) | 7.9 | -1.3 | 7.9 | -0.4 | -0.9 |
Baseline | % Change from Baseline (LSMean 1 ) | Baseline | % Change From Baseline (LSMean 1 ) | Relative Difference of SAXENDA to Placebo (LSMean) | |
Total Cholesterol (mg/dL) • | 171 | -1.4 | 169.4 | 2.4 | -3.7 |
LDL Cholesterol (mg/dL) • | 86.4 | 0.9 | 85.2 | 3.3 | -2.3 |
HDL Cholesterol (mg/dL) • | 45.2 | 4.8 | 45.4 | 1.9 | 2.9 |
Triglycerides (mg/dL) † | 156.2 | -14.5 | 155.8 | -0.7 | -13.5 |
Based on last observation carried forward method while on study drug
1 Least squares mean adjusted for treatment, country, sex, background treatment, baseline HbA 1c stratum and an interaction between background treatment and HbA 1c stratum as fixed factors, and the baseline value as covariate.
2 See Warnings and Precautions (5.5 )
• Baseline value is the geometric mean
† Values are baseline median, median % change, and the Hodges-Lehmann estimate of the median treatment difference.
Weight Management Trial in Pediatric Patients Ages 12 and Older with Obesity
SAXENDA was evaluated in a 56-week, double-blind, randomized, parallel group, placebo controlled multi-center trial in 251 pubertal pediatric patients aged 12 to 17 years, with BMI corresponding to 30 kg/m 2 or greater for adults by international cut-off points 1 and BMI of 95 th percentile or greater for age and sex (NCT02918279). After a 12-week lifestyle run-in period, patients were randomized 1:1 to SAXENDA once-daily or placebo once-daily. The SAXENDA dose was titrated to 3 mg over a 4- to 8-week period based on tolerability as judged by the investigator. Escalation of the trial product was not allowed if the subject had a self-monitored plasma glucose (SMPG) <56 mg/dL or <70 mg/dL in the presence of symptoms of hypoglycemia during the week prior to or during the dose escalation visits. The proportion of patients who reached the 3 mg dose was 82.4%; for 8.8% of patients 2.4 mg was the maximum tolerated dose.
The mean age was 14.5 years: 40.6% of patients were male, 87.6% were White, 0.8% were Asian, 8% were Black or African American; 22.3% were of Hispanic or Latino ethnicity. The mean baseline body weight was 100.8 kg, and mean Body Mass Index (BMI) was 35.6 kg/m 2 .
The proportions of patients who discontinued study drug were 19.2% for the SAXENDA-treated group and 20.6% for the placebo-treated group; 10.4% of patients treated with SAXENDA and no patients treated with placebo discontinued treatment due to an adverse reaction [see Adverse Reactions (6.1 )] .
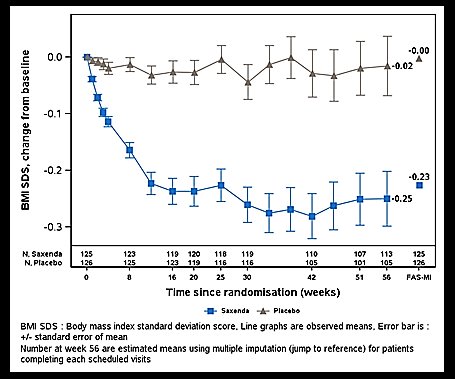
The primary endpoint was change in BMI SDS. At baseline, mean BMI SDS was 3.14 in the SAXENDA group and 3.20 in the placebo group. At Week 56, treatment with SAXENDA resulted in statistically significant reduction in BMI SDS from baseline compared to placebo. The observed mean change in BMI SDS from baseline to Week 56 was -0.23 in the SAXENDA group and -0.00 in the placebo group. The estimated treatment difference in BMI SDS reduction from baseline between SAXENDA and placebo was -0.22 with a 95% confidence interval of -0.37, -0.08; p=0.0022.
The time course of change in BMI SDS with SAXENDA and placebo from baseline through Week 56 are depicted in Figure 5 .
Figure 5. Change from Baseline in BMI SDS
Changes in weight and BMI with SAXENDA are shown in Table 8 . Changes in waist circumference and cardiometabolic parameters with SAXENDA are shown in Table 9 .
Table 8. Changes in Weight and BMI at Week 56 for Study 4 (Pediatric Patients Ages 12 to Less than 18)
SAXENDA N=125 | Placebo N=126 | SAXENDA minus Placebo | |
Body Weight | |||
| 99.3 | 102.2 | |
| -2.65 | 2.37 | -5.01 |
BMI | |||
| 35.3 | 35.8 | |
| -4.29 | 0.35 | -4.64 |
Proportion of patients with greater than or equal to 5% reduction in baseline BMI at Week 56 (%) | 43.3% | 18.7% | 24.6% |
Proportion of patients with greater than or equal to 10% reduction in baseline BMI at Week 56 (%) | 26.1% | 8.1% | 18% |
Full Analysis Set. For body weight and BMI, baseline values are means, changes from baseline at Week 56 are estimated means (least-squares) and treatment contrasts at Week 56 are estimated treatment differences. Missing observations were imputed from the placebo arm based on a jump to reference multiple (x100) imputation approach.
Table 9. Mean Changes in Anthropometry and Cardiometabolic Parameters in Study 4 (Pediatric Patients Ages 12 to Less than 18)
SAXENDA N=125 | Placebo N=126 | ||||
Baseline | Change from Baseline | Baseline | Change from Baseline | SAXENDA minus Placebo | |
Waist Circumference (cm) | 105 | -4.35 | 107 | -1.42 | -2.93 |
Systolic Blood Pressure (mmHg) | 116 | -1.21 | 117 | 0.84 | -2.05 |
Diastolic Blood Pressure (mmHg) | 72 | 0.77 | 73 | -0.46 | 1.24 |
Heart Rate (bpm) • | 75 | 1.87 | 78 | -0.14 | 2.01 |
HbA 1c (%) | 5.3 | -0.1 | 5.3 | -0.03 | -0.06 |
Baseline | % Change from Baseline | Baseline | % Change from Baseline | Relative Difference of SAXENDA to Placebo | |
Total Cholesterol (mg/dL) •• | 154.2 | 0.84 | 152.2 | -0.03 | 0.88 |
LDL Cholesterol (mg/dL) •• | 85.5 | 1.74 | 82.5 | 3.01 | -1.27 |
HDL Cholesterol (mg/dL) •• | 42.7 | 5.14 | 42.7 | 3.33 | 1.81 |
Triglycerides (mg/dL) •• | 109.1 | -0.12 | 112.2 | -1.35 | 1.23 |
• See Warnings and Precautions (5.5 ) | |||||
Full Analysis Set. Baseline values are means, changes from baseline at Week 56 are estimated means (least-squares) and treatment contrasts at Week 56 are estimated treatment differences. Missing observations were imputed from the placebo arm based on a jump to reference multiple (x100) imputation approach.
- •• Baseline values are geometric means.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
SAXENDA injection: 6 mg/mL clear, colorless solution in a 3 mL single-patient-use prefilled pen that delivers doses of 0.6 mg, 1.2 mg, 1.8 mg, 2.4 mg or 3 mg is available in the following package sizes:
3 x SAXENDA pen NDC 0169-2800-13
5 x SAXENDA pen NDC 0169-2800-15
Recommended Storage
Prior to first use, SAXENDA should be stored in a refrigerator between 36ºF to 46ºF (2ºC to 8ºC). Do not store in the freezer or directly adjacent to the refrigerator cooling element. Do not freeze SAXENDA and do not use SAXENDA if it has been frozen.
After initial use of the SAXENDA pen, the pen can be stored for 30 days at controlled room temperature 59°F to 86°F (15°C to 30°C) or in a refrigerator 36°F to 46°F (2°C to 8°C). Keep the pen cap on when not in use. Protect SAXENDA from excessive heat and sunlight. Always remove and safely discard the needle after each injection and store the SAXENDA pen without an injection needle attached. This will reduce the potential for contamination, infection, and leakage while also ensuring dosing accuracy.
Instructions for Use
- Instructions for Use
- Read these instructions carefully before using your Saxenda ® pen.
- Do not use your pen without proper training from your healthcare provider. Make sure that you know how to give yourself an injection with the pen before you start your treatment.
 If you are blind or have poor eyesight and cannot read the dose counter on the pen, do not use this pen without help. Get help from a person with good eyesight who is trained to use the Saxenda pen .
If you are blind or have poor eyesight and cannot read the dose counter on the pen, do not use this pen without help. Get help from a person with good eyesight who is trained to use the Saxenda pen . - You can refresh your training at any time by watching the online training video at www.saxenda.com .
- Start by checking your pen to make sure that it contains Saxenda, then look at the pictures below to get to know the different parts of your pen and needle.
- Your pen is a prefilled, dial-a-dose, single-patient-use pen. It contains 18 mg of liraglutide, and you can select doses of 0.6 mg, 1.2 mg, 1.8 mg, 2.4 mg and 3 mg. Your pen is made to be used with disposable needles up to a length of 8 mm. Pen needles are not included with your Saxenda pen.
- Supplies you will need to give your Saxenda injection:
- Saxenda pen
- a new disposable needle
- alcohol swab
- cotton ball or gauze
- sharps disposal container for throwing away used Saxenda pens and needles.
See “Always dispose of the needle after each injection” at the end of these instructions.
Step 1. Prepare your pen with a new needle
|  |
|  |
| 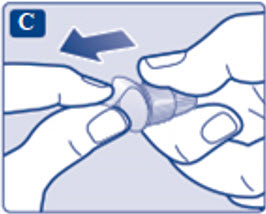 |
| 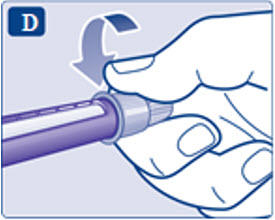 |
| 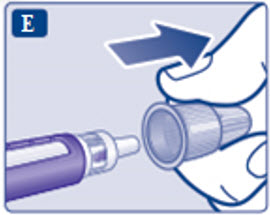 |
| 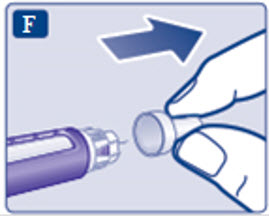 |
Step 2. Check the Saxenda flow with each new pen.
| 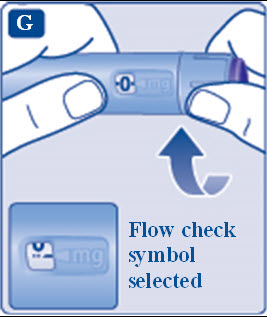 |
| 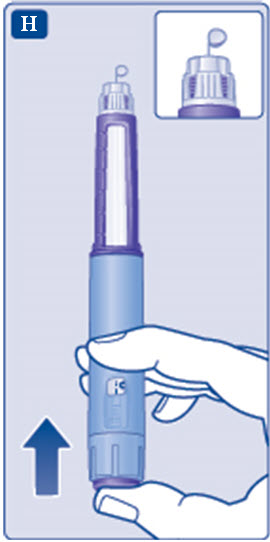 |
Step 3. Select your dose
| 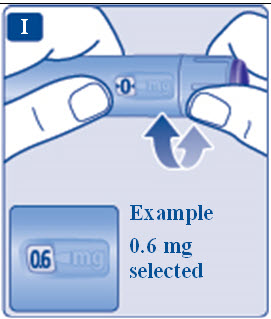 |
| 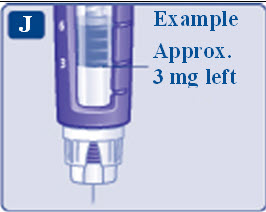 |
| 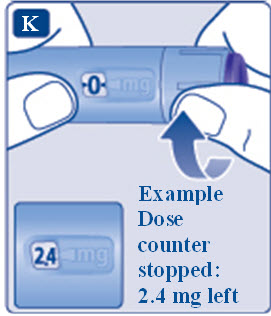 |
Step 4. Inject your dose
| 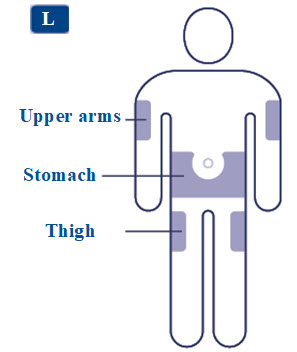  |
|  |
| 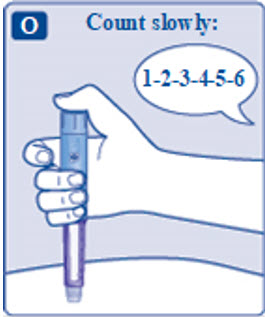 |
| 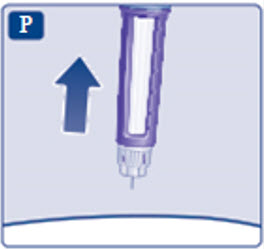 |
Step 5. After your injection
| 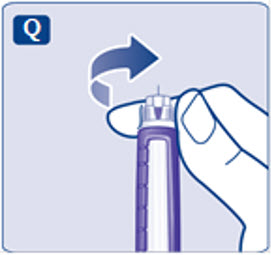 |
|  |
| 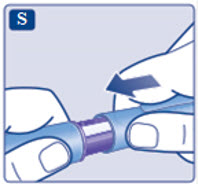 |
| 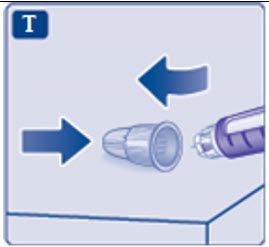 |
| |
Caring for your pen
How should I store my Saxenda pen?
| |
 Always use a new needle for each injection.
Always use a new needle for each injection.  Do not attach a new needle to your pen until you are ready to take your injection.
Do not attach a new needle to your pen until you are ready to take your injection.  ) .
) .  Always make sure that a drop appears at the needle tip before you use a new pen for the first time. This makes sure that Saxenda flows.
Always make sure that a drop appears at the needle tip before you use a new pen for the first time. This makes sure that Saxenda flows.  A small drop may remain at the needle tip, but it will not be injected.
A small drop may remain at the needle tip, but it will not be injected.  Always use the dose counter and the dose pointer to see how many mg you select.
Always use the dose counter and the dose pointer to see how many mg you select.  Only doses of 0.6 mg, 1.2 mg, 1.8 mg, 2.4 mg or 3 mg can be selected with the dose selector. The selected dose must line up exactly with the dose pointer to make sure that you get a correct dose.
Only doses of 0.6 mg, 1.2 mg, 1.8 mg, 2.4 mg or 3 mg can be selected with the dose selector. The selected dose must line up exactly with the dose pointer to make sure that you get a correct dose.  How much Saxenda is left?
How much Saxenda is left?  If you need more Saxenda than what is left in your pen
If you need more Saxenda than what is left in your pen  Be very careful to calculate correctly.
Be very careful to calculate correctly.  If blood appears at the injection site, press lightly with a cotton ball or gauze. Do not rub the area.
If blood appears at the injection site, press lightly with a cotton ball or gauze. Do not rub the area.  Never touch the dose counter when you inject. This can stop the injection.
Never touch the dose counter when you inject. This can stop the injection.  If you do not have a sharps container, follow a 1-handed needle recapping method. Carefully slip the needle into the outer needle cap. Dispose of the needle in a sharps container as soon as possible.
If you do not have a sharps container, follow a 1-handed needle recapping method. Carefully slip the needle into the outer needle cap. Dispose of the needle in a sharps container as soon as possible.  Never try to put the inner needle cap back on the needle.
Never try to put the inner needle cap back on the needle.  Always dispose of the needle after each injection .
Always dispose of the needle after each injection .  Important
Important - This Instructions for Use have been approved by the U.S. Food and Drug Administration. Revised: 06/2026
Mechanism of Action
Liraglutide is an acylated human GLP-1 receptor agonist with 97% amino acid sequence homology to endogenous human GLP-1(7-37). Like endogenous GLP-1, liraglutide binds to and activates the GLP-1 receptor, a cell-surface receptor coupled to adenylyl cyclase activation through the stimulatory G-protein, Gs. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the ubiquitous endogenous enzymes, dipeptidyl peptidase 4 (DPP-4) and neutral endopeptidases (NEP). Unlike native GLP-1, liraglutide is stable against metabolic degradation by both peptidases and has a plasma half-life of 13 hours after subcutaneous administration. The pharmacokinetic profile of liraglutide, which makes it suitable for once-daily administration, is a result of self-association that delays absorption, plasma protein binding, and stability against metabolic degradation by DPP-4 and NEP.
GLP-1 is a physiological regulator of appetite and calorie intake, and the GLP-1 receptor is present in several areas of the brain involved in appetite regulation. In animal studies, peripheral administration of liraglutide resulted in the presence of liraglutide in specific brain regions regulating appetite, including the hypothalamus. Although liraglutide activated neurons in brain regions known to regulate appetite, specific brain regions mediating the effects of liraglutide on appetite were not identified in rats.