Get your patient on Taltz (Ixekizumab)
Patient education
Administration guides
Patient education materials
Treatment initiation and patient onboarding
Patient support program
Dosing resources
Clinical information
Insurance resources
Prior authorization & coverage support
Reimbursement information
Financial assistance & copay programs
Specialty pharmacy coordination
Other resources
Dosage & administration
Coverage
See specific coverage requirements, including prior authorization and step therapies.

Taltz prescribing information
INDICATIONS AND USAGE
TALTZ ® is a humanized interleukin-17A antagonist indicated for the treatment of:
- patients aged 6 years or older with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. (1.1 )
- adults with active psoriatic arthritis. (1.2 )
- adults with active ankylosing spondylitis. (1.3 )
- adults with active non-radiographic axial spondyloarthritis with objective signs of inflammation. (1.4 )
Plaque Psoriasis
TALTZ ® is indicated for the treatment of patients 6 years of age and older with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
Psoriatic Arthritis
TALTZ is indicated for the treatment of adult patients with active psoriatic arthritis.
Ankylosing Spondylitis
TALTZ is indicated for the treatment of adult patients with active ankylosing spondylitis.
Non-radiographic Axial Spondyloarthritis
TALTZ is indicated for the treatment of adult patients with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation.
DOSAGE AND ADMINISTRATION
Administer by subcutaneous injection.
Adult Plaque Psoriasis (2.2 )
- Recommended dosage is 160 mg (two 80 mg injections) at Week 0, followed by 80 mg at Weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeks.
Pediatric Plaque Psoriasis (2.3 )
- For patients weighing greater than 50 kg, recommended dosage is 160 mg (two 80 mg injections) at Week 0, followed by 80 mg every 4 weeks.
- For patients weighing 25-50 kg, recommended dosage is 80 mg at Week 0, followed by 40 mg every 4 weeks.
- For patients weighing less than 25 kg, recommended dosage is 40 mg at Week 0, followed by 20 mg every 4 weeks.
Psoriatic Arthritis (2.4 )
- Recommended dosage is 160 mg by subcutaneous injection (two 80 mg injections) at Week 0, followed by 80 mg every 4 weeks.
- For psoriatic arthritis patients with coexistent moderate-to-severe plaque psoriasis, use the dosing regimen for adult plaque psoriasis. (2.2 )
- TALTZ may be administered alone or in combination with a conventional DMARD (e.g., methotrexate).
Ankylosing Spondylitis (2.5 )
- Recommended dosage is 160 mg by subcutaneous injection (two 80 mg injections) at Week 0, followed by 80 mg every 4 weeks.
Non-radiographic Axial Spondyloarthritis (2.6 )
- Recommended dosage is 80 mg by subcutaneous injection every 4 weeks.
Testing and Procedures Prior to Treatment Initiation
Perform the following evaluations prior to TALTZ initiation:
- Evaluate patients for tuberculosis (TB) infection. TALTZ initiation is not recommended in patients with active TB infection. Initiate treatment of latent TB prior to initiation of TALTZ [see Warnings and Precautions (5.2 )] .
- Complete all age-appropriate vaccinations as recommended by current immunization guidelines prior to initiating treatment with TALTZ [see Warnings and Precautions (5.6 )] .
Recommended Dosage in Adult Plaque Psoriasis
TALTZ is administered by subcutaneous injection. The recommended dosage in adults with moderate-to-severe plaque psoriasis is 160 mg (two 80 mg injections) at Week 0, followed by 80 mg at Weeks 2, 4, 6, 8, 10, and 12, then 80 mg every 4 weeks.
Recommended Dosage in Pediatric Plaque Psoriasis
TALTZ is administered by subcutaneous injection every 4 weeks (Q4W). The recommended dosage in pediatric patients from 6 to less than 18 years of age with moderate-to-severe plaque psoriasis is based on the following weight categories.
| Pediatric Patient's Weight | Starting Dose (Week 0) | Dose every 4 weeks (Q4W) Thereafter |
| Greater than 50 kg | 160 mg (two 80 mg injections) | 80 mg |
| 25 to 50 kg | 80 mg | 40 mg |
| Less than 25 kg | 40 mg | 20 mg |
Recommended Dosage in Psoriatic Arthritis
The recommended dosage is 160 mg by subcutaneous injection (two 80 mg injections) at Week 0, followed by 80 mg every 4 weeks.
For psoriatic arthritis patients with coexistent moderate-to-severe plaque psoriasis, use the dosing regimen for adult plaque psoriasis [see Dosage and Administration (2.2 )] .
TALTZ may be administered alone or in combination with a conventional disease-modifying antirheumatic drug (cDMARD) (e.g., methotrexate).
Recommended Dosage in Ankylosing Spondylitis
The recommended dosage is 160 mg by subcutaneous injection (two 80 mg injections) at Week 0, followed by 80 mg every 4 weeks.
Recommended Dosage in Non-radiographic Axial Spondyloarthritis
The recommended dosage is 80 mg by subcutaneous injection every 4 weeks.
Preparation and Administration Instructions
TALTZ is intended for use under the guidance and supervision of a healthcare provider. Adult patients may self-inject or caregivers may give injections of TALTZ after training in subcutaneous injection technique using the autoinjector or prefilled syringe. Safety and effectiveness of pediatric self-administration has not been established. Therefore, TALTZ should be administered to pediatric patients by a healthcare provider or by a caregiver who has received training and demonstrated proper subcutaneous injection technique.
TALTZ 20 mg and 40 mg doses prepared from the TALTZ 80 mg/mL prefilled syringe should only be administered by a qualified healthcare professional [see Dosage and Administration (2.8 )] .
The TALTZ “Instructions for Use” contains more detailed instructions on the preparation and administration of TALTZ [see Instructions for Use] .
Before injection, remove TALTZ autoinjector or TALTZ prefilled syringe from the refrigerator and allow TALTZ to reach room temperature (30 minutes) without removing the needle cap. Inspect TALTZ visually for particulate matter and discoloration prior to administration. TALTZ is a clear and colorless to slightly yellow solution. Do not use if the liquid contains visible particles, is discolored or cloudy (other than clear and colorless to slightly yellow).
Administer each injection at a different anatomic location (such as upper arms, thighs or any quadrant of abdomen) than the previous injection, and not into areas where the skin is tender, bruised, erythematous, indurated or affected by psoriasis. Administration of TALTZ in the upper, outer arm may be performed by a caregiver or healthcare provider [see Instructions for Use] .
TALTZ does not contain preservatives, therefore discard any unused product.
If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
Alternative Preparation Instructions of TALTZ Doses for Pediatric Patients with Plaque Psoriasis Weighing 50 kg or Less
If the 20 mg/0.25 mL or 40 mg/0.5 mL prefilled syringe is unavailable, TALTZ doses of 20 mg or 40 mg for pediatric patients with plaque psoriasis [see Dosage and Administration (2.3 )] must be manually prepared according to the steps below using only the TALTZ 80 mg/mL prefilled syringe. The preparation and administration of the 20 mg and 40 mg doses should only be performed by a qualified healthcare professional. For additional preparation and administration instructions, [see Dosage and Administration (2.7 )] .
- Gather the following necessary supplies for preparation:
- 0.5 mL or 1 mL disposable syringe
- Sterile needle for withdrawal
- 27-gauge sterile needle for administration
- Sterile, clear glass vial.
- Expel the entire contents of the prefilled syringe into the sterile vial. DO NOT shake or swirl the vial. Do not add other medications to solutions containing TALTZ.
- Using the 0.5 mL or 1 mL disposable syringe and sterile needle, withdraw the prescribed dose from the vial (0.25 mL for 20 mg; 0.5 mL for 40 mg).
- Remove the needle from the syringe and replace it with a 27-gauge needle prior to administering TALTZ.
Storage of Prepared TALTZ
If necessary, TALTZ 20 mg or 40 mg doses prepared from an 80 mg/mL prefilled syringe may be stored at room temperature for up to 4 hours from first puncturing the sterile vial.
DOSAGE FORMS AND STRENGTHS
TALTZ is a clear and colorless to slightly yellow solution available as:
- Injection: 80 mg/mL solution of TALTZ in a single-dose prefilled autoinjector
- Injection: 80 mg/mL solution of TALTZ in a single-dose prefilled syringe
- Injection: 40 mg/0.5 mL solution of TALTZ in a single-dose prefilled syringe
- Injection: 20 mg/0.25 mL solution of TALTZ in a single-dose prefilled syringe
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to TALTZ during pregnancy. Pregnant women exposed to TALTZ are encouraged to enroll in the TALTZ Pregnancy Registry by calling 1-800-284-1695. Contact information for the registry is also available on http://www.pregnancyregistry.lilly.com.
Risk Summary
Available data from the published literature and the pharmacovigilance database with TALTZ use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes.
Human IgG is known to cross the placental barrier; therefore, TALTZ may be transmitted from the mother to the developing fetus. An embryofetal development study conducted in pregnant monkeys during organogenesis at doses up to 19 times the maximum recommended human dose (MRHD) revealed no evidence of harm to the developing fetus. When dosing was continued until parturition, neonatal deaths were observed at 1.9 times the MRHD [see Data] . The clinical significance of these nonclinical findings is unknown.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
An embryofetal development study was conducted in cynomolgus monkeys administered ixekizumab. No malformations or embryofetal toxicity were observed in fetuses from pregnant monkeys administered ixekizumab weekly by subcutaneous injection during organogenesis to near parturition at doses up to 19 times the MRHD (on a mg/kg basis of 50 mg/kg/week). Ixekizumab crossed the placenta in monkeys.
In a pre- and post-natal development toxicity study, pregnant cynomolgus monkeys were administered weekly subcutaneous doses of ixekizumab up to 19 times the MRHD from the beginning of organogenesis to parturition. Neonatal deaths occurred in the offspring of two monkeys administered ixekizumab at 1.9 times the MRHD (on a mg/kg basis of 5 mg/kg/week) and two monkeys administered ixekizumab at 19 times the MRHD (on a mg/kg basis of 50 mg/kg/week). These neonatal deaths were attributed to early delivery, trauma, or congenital defect. The clinical significance of these findings is unknown. No ixekizumab-related effects on functional or immunological development were observed in the surviving infants from birth through 6 months of age.
Lactation
Risk Summary
There are no available data on the presence of ixekizumab in human milk, the effects on the breastfed infant, or the effects on milk production. Ixekizumab was detected in the milk of lactating cynomolgus monkeys. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TALTZ and any potential adverse effects on the breastfed infant from TALTZ or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of TALTZ have been established in pediatric subjects aged 6 years to less than 18 years with moderate-to-severe plaque psoriasis. The safety and effectiveness of TALTZ in other pediatric indications and for pediatric subjects less than 6 years of age have not been established.
Geriatric Use
Of the 4204 adult psoriasis subjects exposed to TALTZ, a total of 301 were 65 years or older, and 36 subjects were 75 years or older. Although no differences in safety or efficacy were observed between older and younger subjects, the number of subjects aged 65 and over is not sufficient to determine whether they respond differently from younger subjects [see Clinical Pharmacology (12.3 )] .
CONTRAINDICATIONS
TALTZ is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients [see Warnings and Precautions (5.3 )] .
WARNINGS AND PRECAUTIONS
- Infections : Serious infections have occurred. Instruct patients to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a serious infection develops, discontinue TALTZ until the infection resolves. (5.1 )
- Tuberculosis (TB) : Evaluate for TB prior to initiating treatment. (5.2 )
- Hypersensitivity : If a serious allergic reaction occurs, discontinue TALTZ immediately and initiate appropriate therapy. (5.3 )
- Eczematous Eruptions: In the postmarketing setting, cases of severe eczematous eruptions were reported in patients receiving TALTZ. Treatment may need to be discontinued to resolve the eczematous eruption. (5.4 )
- Inflammatory Bowel Disease : Crohn's disease and ulcerative colitis, including exacerbations, occurred during clinical trials. Monitor closely when prescribing TALTZ to patients with inflammatory bowel disease (IBD). Discontinue TALTZ and initiate appropriate medical management if IBD develops. (5.5 )
- Immunizations : Avoid use of live vaccines. (5.6 )
Infections
TALTZ may increase the risk of infection. In clinical trials in adult patients with plaque psoriasis, the TALTZ group had a higher rate of infections than the placebo group (27% vs. 23%). Upper respiratory tract infections, oral candidiasis, conjunctivitis and tinea infections occurred more frequently in the TALTZ group than in the placebo group. A similar increase in risk of infection was seen in placebo-controlled trials in patients with pediatric psoriasis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis [see Adverse Reactions (6.1 )] .
In the postmarketing setting, serious bacterial, viral, and fungal opportunistic infections have been reported in patients receiving IL-17 inhibitors including TALTZ. Instruct patients treated with TALTZ to seek medical advice if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a serious infection or is not responding to standard therapy, monitor the patient closely and discontinue TALTZ until the infection resolves.
Pre-treatment Evaluation for Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with TALTZ. Do not administer to patients with active TB infection. Initiate treatment of latent TB prior to administering TALTZ. Consider anti-TB therapy prior to initiating TALTZ in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Patients receiving TALTZ should be monitored closely for signs and symptoms of active TB during and after treatment.
Hypersensitivity
Serious hypersensitivity reactions, including angioedema and urticaria (each ≤0.1%), occurred in the TALTZ group in clinical trials. Anaphylaxis, including cases leading to hospitalization, has been reported in post marketing use with TALTZ [see Adverse Reactions (6.1 , 6.3 )] . If a serious hypersensitivity reaction occurs, discontinue TALTZ immediately and initiate appropriate therapy.
Eczematous Eruptions
In the postmarketing setting, cases of severe eczematous eruptions, including atopic dermatitis-like eruptions, dyshidrotic eczema, and erythroderma were reported in patients receiving TALTZ; some cases resulted in hospitalization. The onset of eczematous eruptions was variable, ranging from days to months after the first dose of TALTZ. Treatment may need to be discontinued to resolve the eczematous eruption. Some patients with limited psoriasis treatment options were successfully treated for eczema while continuing TALTZ.
Inflammatory Bowel Disease
Patients treated with TALTZ may be at increased risk of inflammatory bowel disease. In clinical trials, Crohn's disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the TALTZ group than the placebo control group [see Adverse Reactions (6.1 )] . During TALTZ treatment, monitor for onset or exacerbation of inflammatory bowel disease and if IBD occurs, discontinue TALTZ and initiate appropriate medical management.
Immunizations
Prior to initiating therapy with TALTZ, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with TALTZ. No data are available on the response to live vaccines.
ADVERSE REACTIONS
The following adverse drug reactions are discussed in greater detail in other sections of the label:
Clinical Trials Experience
Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Plaque Psoriasis
Weeks 0 to 12 :
Three placebo-controlled trials in subjects with plaque psoriasis were integrated to evaluate the safety of TALTZ compared to placebo for up to 12 weeks. A total of 1167 subjects (mean age 45 years; 66% men; 94% White) with plaque psoriasis received TALTZ (160 mg at Week 0, 80 mg every 2 weeks [Q2W] for 12 weeks) subcutaneously. In two of the trials, the safety of TALTZ (use up to 12 weeks) was also compared with an active comparator, U.S. approved etanercept [see Clinical Studies (14 )] .
In the 12-week, placebo-controlled period, adverse events occurred in 58% of the TALTZ Q2W group (2.5 per subject-year of follow-up) compared with 47% of the placebo group (2.1 per subject-year of follow-up). Serious adverse events occurred in 2% of the TALTZ group (0.07 per subject-year of follow-up), and in 2% of the placebo group (0.07 per subject-year of follow-up).
Table 2 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the TALTZ group than in the placebo group during the 12-week placebo-controlled period of the pooled clinical trials.
a Upper respiratory tract infections cluster includes nasopharyngitis and rhinovirus infection. | |||
b U.S. approved etanercept. | |||
| Adverse Reactions | TALTZ 80 mg Q2W (N=1167) n (%) | Etanercept b (N=287) n (%) | Placebo (N=791) n (%) |
| Injection site reactions | 196 (17) | 32 (11) | 26 (3) |
| Upper respiratory tract infections a | 163 (14) | 23 (8) | 101 (13) |
| Nausea | 23 (2) | 1 (<1) | 5 (1) |
| Tinea infections | 17 (2) | 0 | 1 (<1) |
Adverse reactions that occurred at rates less than 1% in the TALTZ group and more frequently than in the placebo group during the 12-week induction period included rhinitis, oral candidiasis, urticaria, influenza, conjunctivitis, inflammatory bowel disease, and angioedema.
Weeks 13 to 60 :
A total of 332 subjects received the recommended maintenance regimen of TALTZ 80 mg dosed every 4 weeks.
During the maintenance period (Weeks 13 to 60), adverse events occurred in 80% of subjects treated with TALTZ (1.0 per subject-year of follow-up) compared to 58% of subjects treated with placebo (1.1 per subject-year of follow-up). Serious adverse events were reported in 4% of subjects treated with TALTZ (0.05 per subject-year of follow-up) and none in the subjects treated with placebo.
Weeks 0 to 60 :
Over the entire treatment period (Weeks 0 to 60), adverse events were reported in 67% of subjects treated with TALTZ (1.4 per subject-year of follow-up) compared to 48% of subjects treated with placebo (2.0 per subject-year of follow-up). Serious adverse events were reported in 3% of subjects treated with TALTZ (0.06 per subject-year of follow-up), and in 2% of subjects treated with placebo (0.06 per subject-year of follow-up).
Specific Adverse Drug Reactions
Injection Site Reactions
The most frequent injection site reactions were erythema and pain. Most injection site reactions were mild-to-moderate in severity and did not lead to discontinuation of TALTZ.
Infections
In the 12-week, placebo-controlled period of the clinical trials in plaque psoriasis, infections occurred in 27% of subjects treated with TALTZ (1.2 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.4% of subjects treated with TALTZ (0.02 per subject-year of follow-up) and in 0.4% of subjects treated with placebo (0.02 per subject-year of follow-up) [see Warnings and Precautions (5.1 )] .
During the maintenance treatment period (Weeks 13 to 60), infections occurred in 57% of subjects treated with TALTZ (0.70 per subject-year of follow-up) compared to 32% of subjects treated with placebo (0.61 per subject-year of follow-up). Serious infections occurred in 0.9% of subjects treated with TALTZ (0.01 per subject-year of follow-up) and none in the subjects treated with placebo.
Over the entire treatment period (Weeks 0 to 60), infections were reported in 38% of subjects treated with TALTZ (0.83 per subject-year of follow-up) compared to 23% of subjects treated with placebo (1.0 per subject-year of follow-up). Serious infections occurred in 0.7% of subjects treated with TALTZ (0.02 per subject-year of follow-up), and in 0.4% of subject treated with placebo (0.02 per subject-year of follow-up).
Inflammatory Bowel Disease
In adult subjects with plaque psoriasis, Crohn's disease and ulcerative colitis, including exacerbations, occurred at a greater frequency in the TALTZ 80 mg Q2W group (Crohn's disease 0.1%, ulcerative colitis 0.2%) than the placebo group (0%) during the 12-week, placebo-controlled period in clinical trials [see Warnings and Precautions (5.5 )] .
Laboratory Assessment of Cytopenia
Neutropenia
Over the entire treatment period (Weeks 0 to 60), neutropenia occurred in 11% of subjects treated with TALTZ (0.24 per subject-year of follow-up) compared to 3% of subjects treated with placebo (0.14 per subject-year of follow-up). In subjects treated with TALTZ, the incidence rate of neutropenia during Weeks 13 to 60 was lower than the incidence rate during Weeks 0 to 12.
In the 12-week, placebo-controlled period, neutropenia ≥ Grade 3 (<1,000 cells/mm 3 ) occurred in 0.2% of the TALTZ group (0.007 per subject-year of follow-up) compared to 0.1% of the placebo group (0.006 per subject-year of follow-up). The majority of cases of neutropenia were either Grade 2 (2% for TALTZ 80 mg Q2W versus 0.3% for placebo; ≥1,000 to <1,500 cells/mm 3 ) or Grade 1 (7% for TALTZ 80 mg Q2W versus 3% for placebo; ≥1,500 cells/mm 3 to ˂2,000 cells/mm 3 ). Neutropenia in the TALTZ group was not associated with an increased rate of infection compared to the placebo group.
Thrombocytopenia
Ninety eight percent of cases of thrombocytopenia were Grade 1 (3% for TALTZ 80 mg Q2W versus 1% for placebo; ≥75,000 cells/mm 3 to <150,000 cells/mm 3 ). Thrombocytopenia in subjects treated with TALTZ was not associated with an increased rate of bleeding compared to subjects treated with placebo.
Active Comparator Trials
In the two clinical trials that included an active comparator, the rate of serious adverse events during weeks zero to twelve was 0.7% for U.S. approved etanercept and 2% for TALTZ 80 mg Q2W, and the rate of discontinuation from adverse events was 0.7% for U.S. approved etanercept and 2% for TALTZ 80 mg Q2W. The incidence of infections was 18% for U.S. approved etanercept and 26% for TALTZ 80 mg Q2W. The rate of serious infections was 0.3% for both TALTZ 80 mg Q2W and U.S. approved etanercept.
Pediatric Plaque Psoriasis
TALTZ was evaluated in a placebo-controlled trial in pediatric subjects with moderate-to-severe psoriasis 6 to less than 18 years of age. A total of 171 subjects were studied (115 subjects on TALTZ and 56 subjects on placebo). Overall, the safety profile observed in pediatric subjects with plaque psoriasis treated with TALTZ every 4 weeks is consistent with the safety profile in adult subjects with plaque psoriasis with the exception of the frequencies of conjunctivitis (2.6%), influenza (1.7%), and urticaria (1.7%).
In this clinical trial, Crohn's disease occurred at a greater frequency in the TALTZ group (0.9%) than the placebo group (0%) during the 12-week, placebo-controlled period. Crohn's disease occurred in a total of 4 TALTZ treated subjects (2.0%) in the clinical trial [see Warnings and Precautions (5.5 )] .
Psoriatic Arthritis
TALTZ was studied in two placebo-controlled trials in patients with psoriatic arthritis. A total of 678 patients were studied (454 patients on TALTZ and 224 on placebo). A total of 229 patients in these trials received TALTZ 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with psoriatic arthritis treated with TALTZ Q4W is consistent with the safety profile in adult patients with plaque psoriasis with the exception of the frequencies of influenza (1.3%) and conjunctivitis (1.3%).
Ankylosing Spondylitis
TALTZ was studied in two placebo-controlled trials in patients with ankylosing spondylitis. A total of 566 patients were studied (376 patients on TALTZ and 190 on placebo). A total of 195 patients in these trials received TALTZ 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with ankylosing spondylitis treated with TALTZ Q4W is consistent with the safety profile in adult patients with plaque psoriasis.
In adult patients with ankylosing spondylitis, Crohn's disease and ulcerative colitis, including exacerbations, occurred in 2 patients (1.0%) and 1 patient (0.5%), respectively, in the TALTZ 80 mg Q4W group and 1 patient (0.5%) and 0%, respectively, in the placebo group during the 16-week, placebo-controlled period in clinical trials. Of these patients, serious events occurred in 1 patient in the TALTZ 80 mg Q4W group and 1 patient in the placebo group [see Warnings and Precautions (5.5 )] .
Non-radiographic Axial Spondyloarthritis
TALTZ was studied in a placebo-controlled trial in patients with non-radiographic axial spondyloarthritis. A total of 303 patients were studied (198 patients on TALTZ and 105 on placebo). A total of 96 patients in this trial received TALTZ 80 or 160 mg at Week 0, followed by 80 mg every 4 weeks (Q4W). Overall, the safety profile observed in patients with non-radiographic axial spondyloarthritis treated with TALTZ 80 mg Q4W up to Week 16 is consistent with the previous experience of TALTZ in other indications.
Immunogenicity
As with all therapeutic proteins there is the potential for immunogenicity with TALTZ. The assay to test for neutralizing antibodies has limitations detecting neutralizing antibodies in the presence of ixekizumab; therefore, the incidence of neutralizing antibodies development could be underestimated.
Plaque Psoriasis Population
By Week 12, approximately 9% of adult subjects treated with TALTZ every 2 weeks developed antibodies to ixekizumab. Approximately 22% of subjects treated with TALTZ at the recommended dosing regimen developed antibodies to ixekizumab during the 60-week treatment period. The clinical effects of antibodies to ixekizumab are dependent on the antibody titer; higher antibody titers were associated with decreasing drug concentration and clinical response.
Of the adult subjects who developed antibodies to ixekizumab during the 60-week treatment period, approximately 10%, which equates to 2% of subjects treated with TALTZ at the recommended dosing regimen, had antibodies that were classified as neutralizing. Neutralizing antibodies were associated with reduced drug concentrations and loss of efficacy.
In pediatric psoriasis subjects treated with ixekizumab at the recommended dosing regimen up to 12 weeks, 21 subjects (18%) developed anti-drug antibodies, 5 subjects (4%) had confirmed neutralizing antibodies associated with low drug concentrations. No conclusive evidence could be obtained on the potential association of neutralizing antibodies and clinical response and/or adverse events due to small number of pediatric subjects in the study.
Psoriatic Arthritis Population
For subjects treated with TALTZ 80 mg every 4 weeks for up to 52 weeks (PsA1), 11% developed anti-drug antibodies, and 8% had confirmed neutralizing antibodies.
Ankylosing Spondylitis Population
For patients treated with TALTZ 80 mg every 4 weeks for up to 16 weeks (AS1, AS2), 5.2% developed anti-drug antibodies, and 1.5% had neutralizing antibodies.
Non-radiographic Axial Spondyloarthritis Population
Of patients treated with TALTZ 80 mg every 4 weeks for up to 52 weeks (nr-axSpA1), 8.9% developed anti-drug antibodies, all of which were low titer. No patient had neutralizing antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to TALTZ across indications or with the incidences of antibodies to other products may be misleading.
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of TALTZ. Because the reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to TALTZ exposure.
Immune system disorders: anaphylaxis [see Contraindications (4 )] .
Infections: bacterial, viral, and fungal opportunistic infections, including cryptococcal meningoencephalitis, esophageal and disseminated mucocutaneous candidiasis, pulmonary tuberculosis, toxoplasmosis, varicella zoster virus reactivation, cytomegalovirus colitis, pulmonary aspergillosis.
Skin and subcutaneous tissue disorders: Eczematous eruptions (erythroderma, atopic dermatitis-like eruptions, and dyshidrotic eczema).
DESCRIPTION
Ixekizumab is a humanized immunoglobulin G subclass 4 (IgG4) monoclonal antibody (mAb) with neutralizing activity against IL-17A. Ixekizumab is produced by recombinant DNA technology in a recombinant mammalian cell line and purified using standard technology for bioprocessing. Ixekizumab is comprised of two identical light chain polypeptides of 219 amino acids each and two identical heavy chain polypeptides of 445 amino acids each and has a molecular weight of 146,158 Daltons for the protein backbone of the molecule.
TALTZ injection is a sterile, preservative free, clear and colorless to slightly yellow solution, for subcutaneous use available as 80 mg of ixekizumab in a 1 mL single-dose prefilled autoinjector or a single-dose prefilled syringe, 40 mg of ixekizumab in a 0.5 mL single-dose prefilled syringe, or 20 mg of ixekizumab in a 0.25 mL single-dose prefilled syringe. The prefilled autoinjector and prefilled syringe each contain a 1 mL glass syringe with a fixed 27 gauge ½ inch needle. The TALTZ 80 mg prefilled autoinjector and prefilled syringe are manufactured to deliver 80 mg of ixekizumab. The TALTZ 40 mg prefilled syringe is manufactured to deliver 40 mg of ixekizumab. The TALTZ 20 mg prefilled syringe is manufactured to deliver 20 mg of ixekizumab.
Each TALTZ 80 mg/mL single-dose autoinjector or TALTZ 80 mg/mL single-dose prefilled syringe is composed of ixekizumab (80 mg); Polysorbate 80, USP (0.3 mg); Sucrose, USP (80 mg); and Water for Injection, USP. Sodium Hydroxide, USP-NF, may have been added to adjust pH. The TALTZ solution has a pH of 5.2 – 6.2.
Each TALTZ 40 mg/0.5 mL single-dose prefilled syringe is composed of ixekizumab (40 mg); Polysorbate 80, USP (0.15 mg); Sucrose, USP (40 mg); and Water for Injection, USP. Sodium Hydroxide, USP-NF, may have been added to adjust pH. The TALTZ solution has a pH of 5.2 – 6.2.
Each TALTZ 20 mg/0.25 mL single-dose prefilled syringe is composed of ixekizumab (20 mg); Polysorbate 80, USP (0.08 mg); Sucrose, USP (20 mg); and Water for Injection, USP. Sodium Hydroxide, USP-NF, may have been added to adjust pH. The TALTZ solution has a pH of 5.2 – 6.2.
CLINICAL PHARMACOLOGY
Mechanism of Action
Ixekizumab is a humanized IgG4 monoclonal antibody that selectively binds with the interleukin 17A (IL-17A) cytokine and inhibits its interaction with the IL-17 receptor. IL-17A is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Ixekizumab inhibits the release of proinflammatory cytokines and chemokines.
Pharmacodynamics
No formal pharmacodynamic studies have been conducted with TALTZ.
Pharmacokinetics
The pharmacokinetic (PK) properties of ixekizumab were similar across the adult plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis indications.
Absorption
Following a single subcutaneous dose of 160 mg in subjects with plaque psoriasis, ixekizumab reached peak mean (±SD) serum concentrations (C max ) of 16.2 ±6.6 mcg/mL by approximately 4 days post dose.
Steady-state concentrations were achieved by Week 8 following the 160 mg starting dose and 80 mg every 2 weeks dosing regimen; the mean ±SD steady-state trough concentration was 9.3 ±5.3 mcg/mL. Steady-state concentrations were achieved approximately 10 weeks after switching from the 80 mg every 2 weeks dosing regimen to the 80 mg every 4 weeks dosing regimen at Week 12. The mean ±SD steady-state trough concentration was 3.5 ±2.5 mcg/mL.
In studies of subjects with plaque psoriasis, ixekizumab bioavailability ranged from 60% to 81% following subcutaneous injection. Administration of ixekizumab via injection in the thigh achieved a higher bioavailability relative to that achieved using other injection sites including the arm and abdomen.
Distribution
The mean (geometric CV%) volume of distribution at steady-state was 7.11 L (29%) in subjects with plaque psoriasis.
Elimination
The metabolic pathway of ixekizumab has not been characterized. As a humanized IgG4 monoclonal antibody ixekizumab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
The mean systemic clearance was 0.39 L/day (37%) and the mean (geometric CV%) half-life was 13 days (40%) in subjects with plaque psoriasis.
Weight
Ixekizumab clearance and volume of distribution increase as body weight increases.
Dose Linearity
Ixekizumab exhibited dose-proportional pharmacokinetics in subjects with plaque psoriasis over a dose range from 5 mg (not the recommended dose) to 160 mg following subcutaneous administration.
Specific Populations
Age:
Geriatric Population
Population pharmacokinetic analysis indicated that age did not significantly influence the clearance of ixekizumab in adult subjects with plaque psoriasis. Subjects who are 65 years or older had a similar ixekizumab clearance as compared to subjects less than 65 years old.
Pediatric Population
Pediatric psoriasis subjects (6 to less than 18 years of age) were administered ixekizumab at the recommended pediatric dosing regimen for 12 weeks. Subjects weighing >50 kg and 25 to 50 kg had a mean ±SD steady-state trough concentration of 3.8 ±2.2 mcg/mL and 3.9 ±2.4 mcg/mL at Week 12, respectively. There were limited PK data (n=2) in subjects weighing <25 kg at Week 12.
Drug Interaction Studies
Population PK data analyses indicated that the clearance of ixekizumab was not impacted by concomitant administration of methotrexate, or by prior exposure to methotrexate or adalimumab in patients with psoriatic arthritis.
Population PK data analyses indicated that the clearance of ixekizumab was not impacted by concomitant administration of oral corticosteroids, NSAIDs, or cDMARDs (sulfasalazine and methotrexate) in patients with ankylosing spondylitis and non-radiographic axial spondyloarthritis.
Cytochrome P450 Substrates
No clinically significant changes in exposure of caffeine (CYP1A2 substrate), warfarin (CYP2C9 substrate), omeprazole (CYP2C19 substrate) or midazolam (CYP3A substrate) were observed in subjects with plaque psoriasis when used concomitantly with a single 160 mg dose of ixekizumab, or multiple doses of 80 mg every 2 weeks. The potential effect of ixekizumab on the CYP2D6 activity cannot be ruled out due to high variability in exposure (approximately ±2-fold) of dextromethorphan and its CYP2D6 metabolite dextrorphan in psoriasis subjects.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies have not been conducted to evaluate the carcinogenic or mutagenic potential of TALTZ. Moreover published literature is mixed on potential effects on malignancy risk due to the inhibition of IL-17A activity, the pharmacological action of TALTZ. Some published literature suggests that IL-17A directly promotes cancer cell invasion, suggesting a potential beneficial effect by TALTZ, whereas other reports indicate IL-17A promotes T-cell mediated tumor rejection, suggesting a potential adverse effect by TALTZ. However, neutralization of IL-17A with TALTZ has not been studied in these models. Depletion of IL-17A with a neutralizing antibody inhibited tumor development in mice, suggesting a potential beneficial effect by TALTZ. The relevance of experimental findings in mouse models for malignancy risk in humans is unknown.
No effects on fertility parameters such as reproductive organs, menstrual cycle length, or sperm analysis were observed in sexually mature cynomolgus monkeys that were administered ixekizumab for 13 weeks at a subcutaneous dose of 50 mg/kg/week (19 times the MRHD on a mg/kg basis). The monkeys were not mated to evaluate fertility.
CLINICAL STUDIES
Adult Plaque Psoriasis
Three multicenter, randomized, double-blind, placebo-controlled trials, Trials 1, 2, and 3 (NCT 01474512, NCT 01597245, NCT 01646177), enrolled a total of 3866 subjects 18 years of age and older with plaque psoriasis who had a minimum body surface area involvement of 10%, a static Physician Global Assessment (sPGA) score of ≥3 in the overall assessment (plaque thickness/induration, erythema, and scaling) of psoriasis on a severity scale of 0 to 5, a Psoriasis Area and Severity Index (PASI) score ≥12, and who were candidates for phototherapy or systemic therapy.
In all three trials, subjects were randomized to either placebo or TALTZ (80 mg every 2 weeks [Q2W]) for 12 weeks, following a 160 mg starting dose. In the two active comparator trials (Trials 2 and 3), subjects were also randomized to receive U.S. approved etanercept 50 mg twice weekly for 12 weeks.
All three trials assessed the changes from baseline to Week 12 in the two co-primary endpoints: 1) PASI 75, the proportion of subjects who achieved at least a 75% reduction in the PASI composite score that takes into consideration both the percentage of body surface area affected and the nature and severity of psoriatic changes (induration, erythema and scaling) within the affected regions, and 2) sPGA of “0” (clear) or “1” (minimal), the proportion of subjects with an sPGA 0 or 1 and at least a 2-point improvement.
Other evaluated outcomes included the proportion of subjects with an sPGA score of 0 (clear), a reduction of at least 90% in PASI (PASI 90), a reduction of 100% in PASI (PASI 100), and an improvement of itch severity as measured by a reduction of at least 4 points on an 11-point itch Numeric Rating Scale.
Subjects in all treatment groups had a median baseline PASI score ranging from approximately 17 to 18. Baseline sPGA score was severe or very severe in 51% of subjects in Trial 1, 50% in Trial 2, and 48% in Trial 3.
Of all subjects, 44% had received prior phototherapy, 49% had received prior conventional systemic therapy, and 26% had received prior biologic therapy for the treatment of psoriasis. Of the subjects who had received prior biologic therapy, 15% had received at least one anti-TNF alpha agent, and 9% had received an anti-IL 12/IL23. A total of 23% of study subjects had a history of psoriatic arthritis.
Clinical Response at Week 12
The results of Trials 1, 2, and 3 are presented in Table 3 .
a Abbreviations: N = number of patients in the intent-to-treat population; NRI = Non-Responder Imputation. | ||||||
b At Week 0, subjects received 160 mg of TALTZ. | ||||||
c Co-primary endpoints. | ||||||
d Itch NRS (≥4 improvement) in subjects with baseline Itch NRS ≥4. The number of ITT subjects with baseline Itch NRS Score ≥4 are as follows: Trial 1, TALTZ n=391, PBO n=374; Trial 2, TALTZ n=303, PBO n=135; Trial 3, TALTZ n=320, PBO n=158. | ||||||
| Trial 1 | Trial 2 | Trial 3 | ||||
| TALTZ 80 mg b Q2W (N=433) n (%) | Placebo (N=431) n (%) | TALTZ 80 mg b Q2W (N=351) n (%) | Placebo (N=168) n (%) | TALTZ 80 mg b Q2W (N=385) n (%) | Placebo (N=193) n (%) | |
| sPGA of “0” (clear) or “1” (minimal) c | 354 (82) | 14 (3) | 292 (83) | 4 (2) | 310 (81) | 13 (7) |
| sPGA of “0” (clear) | 160 (37) | 0 | 147 (42) | 1 (1) | 155 (40) | 0 |
| PASI 75 c | 386 (89) | 17 (4) | 315 (90) | 4 (2) | 336 (87) | 14 (7) |
| PASI 90 | 307 (71) | 2 (1) | 248 (71) | 1 (1) | 262 (68) | 6 (3) |
| PASI 100 | 153 (35) | 0 | 142 (40) | 1 (1) | 145 (38) | 0 |
| Itch NRS (≥4 point improvement) d | 336 (86) | 58 (16) | 258 (85) | 19 (14) | 264 (83) | 33 (21) |
Examination of age, gender, race, body weight, and previous treatment with a biologic did not identify differences in response to TALTZ among these subgroups at Week 12.
An integrated analysis of the U.S. sites in the two active comparator studies using U.S. approved etanercept, TALTZ demonstrated superiority to U.S. approved etanercept (50 mg twice weekly) on sPGA and PASI scores during the 12-week treatment period. The respective response rates for TALTZ 80 mg Q2W and U.S. approved etanercept 50 mg twice weekly were: sPGA of 0 or 1 (73% and 27%); PASI 75 (87% and 41%); sPGA of 0 (34% and 5%); PASI 90 (64% and 18%), and PASI 100 (34% and 4%).
Maintenance and Durability of Response
To evaluate the maintenance and durability of response, subjects originally randomized to TALTZ and who were responders at Week 12 (i.e., sPGA of 0 or 1) in Trial 1 and Trial 2 were re-randomized to an additional 48 weeks of either a maintenance dose of TALTZ 80 mg Q4W (every 4 weeks) or placebo. Non-responders (sPGA >1) at Week 12 and subjects who relapsed (sPGA ≥3) during the maintenance period were placed on TALTZ 80 mg Q4W.
For responders at Week 12, the percentage of subjects who maintained this response (sPGA 0 or 1) at Week 60 (48 weeks following re-randomization) in the integrated trials (Trial 1 and Trial 2) was higher for subjects treated with TALTZ 80 mg Q4W (75%) compared to those treated with placebo (7%).
For responders at Week 12 who were re-randomized to treatment withdrawal (i.e., placebo), the median time to relapse (sPGA ≥3) was 164 days in the integrated trials. Among these subjects, 66% regained a response of at least 0 or 1 on the sPGA within 12 weeks of restarting treatment with TALTZ 80 mg Q4W.
Psoriasis Involving the Genital Area
A randomized, double-blind, placebo-controlled trial (Trial 4) was conducted in 149 adult subjects with plaque psoriasis who had a minimum body surface area (BSA) involvement of 1%, a sPGA score of ≥3 (moderate psoriasis), a sPGA of Genitalia score of ≥3 (moderate psoriasis involving the genital area), who failed to respond to or were intolerant of at least one topical therapy used for treatment of psoriasis affecting the genital area, and who were candidates for phototherapy and/or systemic therapy.
Subjects had a median baseline PASI score of approximately 12. Baseline BSA involvement was at least 10% for approximately 60% of the enrolled subjects. Baseline sPGA of Genitalia score was severe or very severe in approximately 42% of the subjects; baseline sPGA score was severe or very severe in approximately 47% of the subjects.
Subjects randomized to TALTZ received an initial dose of 160 mg followed by 80 mg every 2 weeks for 12 weeks. The trial evaluated the primary endpoint of the proportion of subjects who achieved a “0” (clear) or “1” (minimal) response at Week 12 on sPGA of Genitalia. Other evaluated outcomes at Week 12 included the proportion of subjects who achieved a sPGA score of “0” (clear) or “1” (minimal), improvement of genital itch severity as measured by a reduction of at least 4 points in the 11-point Genital Psoriasis Symptoms Scale (GPSS) score Itch numeric rating scale (NRS), and the patient-perceived impact of psoriasis affecting the genital area on limiting frequency of sexual activity (intercourse or other activities) as measured by the Genital Psoriasis Sexual Frequency Questionnaire (GenPs-SFQ) Item 2 (In the past week how often did your genital psoriasis limit the frequency of your sexual activity?). SFQ Item 2 score ranges from 0 to 4 (0=never, 1=rarely, 2=sometimes, 3=often, 4=always); where higher scores indicate greater limitations on the frequency of sexual activity in the past week.
The results of Trial 4 are presented in Table 4 .
a Abbreviations: NRI = Non-Responder Imputation; GPSS = Genital Psoriasis Symptoms Scale; GenPs-SFQ = Genital Psoriasis Sexual Frequency Questionnaire. | ||
b At Week 0, subjects received 160 mg of TALTZ, followed by 80 mg every 2 weeks for 12 weeks. | ||
| Endpoints | TALTZ 80 mg Q2W b n (%) | Placebo n (%) |
| Number of subjects randomized | N=75 | N=74 |
| sPGA of Genitalia “0” (clear) or “1” (minimal) | 55 (73%) | 6 (8%) |
| sPGA “0” (clear) or “1” (minimal) | 55 (73%) | 2 (3%) |
| Number of subjects with baseline GPSS a Itch NRS Score ≥4 | N=56 | N=51 |
| GPSS Genital Itch (≥4 point improvement) | 31 (55%) | 3 (6%) |
| Number of subjects with baseline GenPs-SFQ a Item 2 Score ≥2 | N=37 | N=42 |
| GenPs-SFQ Item 2 score “0” (never) or “1” (rarely) | 29 (78%) | 9 (21%) |
Pediatric Plaque Psoriasis
A randomized, double-blind, multicenter, placebo-controlled trial (IXORA-Peds, NCT03073200) enrolled 171 pediatric subjects 6 to less than 18 years of age, with moderate-to-severe plaque psoriasis (as defined by a sPGA score ≥3, involving ≥10% of the body surface area, and a PASI score ≥12) who were candidates for phototherapy or systemic therapy or were inadequately controlled on topical therapy.
Subjects were randomized to placebo or TALTZ with dosing stratified by weight.
- <25 kg: 40 mg at Week 0 followed by 20 mg Q4W
- 25 kg to 50 kg: 80 mg at Week 0 followed by 40 mg Q4W
- >50 kg: 160 mg at Week 0 followed by 80 mg Q4W
Response to treatment was assessed at 12 weeks of therapy and was defined by the proportion of subjects who achieved an sPGA score of “0” (clear) or “1” (almost clear) with at least a 2-point improvement from baseline and the proportion of subjects that achieved a reduction in PASI score of at least 75% (PASI 75) from baseline.
Other evaluated outcomes included the proportion of subjects who achieved PASI 90, PASI 100, sPGA of “0” and an improvement of itch severity as measured by a reduction of at least 4 points on an 11-point itch Numeric Rating Scale.
Subjects had a median baseline PASI score of 17 (range 12-49). Baseline sPGA score was severe or very severe in 49%. Of all subjects, 22% had received prior phototherapy and 32% had received prior conventional systemic therapy for the treatment of psoriasis.
Clinical Response
The efficacy results of IXORA-Peds are presented in Table 5.
a Abbreviations: N = Number of subjects in the intent-to-treat population; NRI = Non-Responder Imputation. | ||
b At Week 0, subjects received 160 mg, 80 mg, or 40 mg of TALTZ, followed by 80 mg, 40 mg, or 20 mg every 4 weeks, depending on weight category, for 12 weeks. | ||
c Co-primary endpoints. | ||
d Itch NRS (≥4 improvement) in subjects with baseline Itch NRS ≥4. The number of ITT subjects with baseline Itch NRS Score ≥4 are as follows: TALTZ, n = 83; PBO, n = 40. | ||
| TALTZ b (N=115) n (%) | Placebo (N=56) n (%) | |
| Week 12 | ||
| sPGA “0” (clear) or “1” (minimal) c | 93 (81%) | 6 (11%) |
| sPGA “0” (clear) | 60 (52%) | 1 (2%) |
| PASI 75 c | 102 (89%) | 14 (25%) |
| PASI 90 | 90 (78%) | 3 (5%) |
| PASI 100 | 57 (50%) | 1 (2%) |
| Itch NRS (≥4 point improvement) d | 59 (71%) | 8 (20%) |
| Week 4 | ||
| sPGA “0” (clear) or “1” (minimal) | 55 (48%) | 4 (7%) |
| PASI 75 | 62 (54%) | 5 (9%) |
Psoriatic Arthritis
The safety and efficacy of TALTZ were assessed in 679 patients, in 2 randomized, double-blind, placebo-controlled studies (PsA1 and PsA2) in adult patients, age 18 years and older with active psoriatic arthritis (at least 3 swollen and at least 3 tender joints) despite non-steroidal anti-inflammatory drug (NSAID), corticosteroid or disease modifying anti-rheumatic drug (DMARD) therapy. Patients in these studies had a diagnosis of PsA for at least 6 months across both studies. At baseline, 60% and 23% of the patients had enthesitis and dactylitis, respectively. In PsA2, all patients discontinued previous treatment with anti-TNFα agents due to either inadequate response or intolerance. In addition, approximately 47% of patients from both studies had concomitant methotrexate (MTX) use.
PsA1 Study (NCT 01695239) evaluated 417 biologic-naive patients, who were treated with either TALTZ 160 mg at Week 0 followed by 80 mg every 2 weeks (Q2W) or 4 weeks (Q4W), adalimumab 40 mg every 2 weeks, or placebo. PsA2 Study (NCT 02349295) evaluated 363 anti-TNFα experienced patients, who were treated with TALTZ 160 mg at Week 0 followed by 80 mg every 2 or 4 weeks, or placebo. Patients receiving placebo were re-randomized to receive TALTZ (80 mg every 2 or 4 weeks) at Week 16 or Week 24 based on responder status. The primary endpoint was the percentage of patients achieving an ACR20 response at Week 24.
Clinical Response
In both studies, patients treated with TALTZ 80 mg Q4W demonstrated a greater clinical response including ACR20, ACR50, and ACR70 compared to placebo at Week 24 (Table 6 ). In PsA2, responses were seen regardless of prior anti-TNFα exposure.
a Patients who met escape criteria (less than 20% improvement in tender and swollen joint counts) at Week 16 or had missing data at Week 24 were considered non-responders at Week 24. | ||||||
b Abbreviations: N = number of patients in the intent-to-treat population; NRI = Non-Responder Imputation. | ||||||
c At Week 0, patients received 160 mg of TALTZ. | ||||||
| PsA1 – anti-TNFα naive | PsA2 – anti-TNFα – experienced | |||||
| TALTZ 80 mg c Q4W (N=107) | Placebo (N=106) | Difference from placebo (95% CI) | TALTZ 80 mg c Q4W (N=122) | Placebo (N=118) | Difference from placebo (95% CI) | |
| ACR20 response | ||||||
| Week 12 (%) | 57 | 31 | 26 (13, 39) | 50 | 22 | 28 (16, 40) |
| Week 24 (%) | 58 | 30 | 28 (15, 41) | 53 | 20 | 34 (22, 45) |
| ACR50 response | ||||||
| Week 12 (%) | 34 | 5 | 29 (19, 39) | 31 | 3 | 28 (19, 37) |
| Week 24 (%) | 40 | 15 | 25 (14, 37) | 35 | 5 | 30 (21, 40) |
| ACR70 response | ||||||
| Week 12 (%) | 15 | 0 | 15 (8, 22) | 15 | 2 | 13 (6, 20) |
| Week 24 (%) | 23 | 6 | 18 (9, 27) | 22 | 0 | 22 (15, 30) |
The percentage of patients achieving ACR20 response by visit is shown in Figure 1 .
Figure 1: Percent of Patients Achieving ACR20 Response a in PsA1 Through Week 24

a Patients who met escape criteria (less than 20% improvement in tender and swollen joint counts) at Week 16 or had missing data at Week 24 were considered non-responders at Week 24.
The improvements in the components of the ACR response criteria are shown in Table 7 .
a At Week 0, subjects received 160 mg of TALTZ. | ||||
b Disability Index of the Health Assessment Questionnaire; 0 = best, 3 = worst, measures the patient's ability to perform the following: dress/groom, arise, eat, walk, reach, grip, maintain hygiene, and maintain daily activity. | ||||
| PsA1 | PsA2 | |||
| TALTZ 80 mg a Q4W (N=107) | Placebo (N=106) | TALTZ 80 mg a Q4W (N=122) | Placebo (N=118) | |
| No. of Swollen Joints | ||||
| Baseline | 11.4 | 10.6 | 13.1 | 10.3 |
| Mean Change at Week 12 | -6.2 | -3.2 | -5.8 | -2.6 |
| Mean Change at Week 16 | -6.2 | -3.0 | -7.4 | -2.6 |
| No. of Tender Joints | ||||
| Baseline | 20.5 | 19.2 | 22.0 | 23.0 |
| Mean Change at Week 12 | -10.3 | -3.5 | -9.4 | -5.4 |
| Mean Change at Week 16 | -9.7 | -4.0 | -10.1 | -3.0 |
| Patient's Assessment of Pain | ||||
| Baseline | 60.1 | 58.5 | 63.9 | 63.9 |
| Mean Change at Week 12 | -26.6 | -9.1 | -29.8 | -11.9 |
| Mean Change at Week 16 | -26.1 | -10.6 | -30.1 | -12.3 |
| Patient Global Assessment | ||||
| Baseline | 62.7 | 61.1 | 66.4 | 64.1 |
| Mean Change at Week 12 | -29.7 | -11.1 | -34.5 | -10.7 |
| Mean Change at Week 16 | -30.4 | -13.2 | -35.3 | -15.7 |
| Physician Global Assessment | ||||
| Baseline | 57.6 | 55.9 | 60.3 | 58.9 |
| Mean Change at Week 12 | -34.0 | -16.6 | -34.4 | -15.9 |
| Mean Change at Week 16 | -35.5 | -16.5 | -32.9 | -9.7 |
| Disability Index (HAQ-DI) b | ||||
| Baseline | 1.2 | 1.2 | 1.2 | 1.2 |
| Mean Change at Week 12 | -0.4 | -0.1 | -0.4 | -0.1 |
| Mean Change at Week 16 | -0.4 | -0.1 | -0.5 | -0.1 |
| CRP (mg/L) | ||||
| Baseline | 12.8 | 15.1 | 17.0 | 12.1 |
| Mean Change at Week 12 | -8.8 | -3.2 | -11.4 | -4.3 |
| Mean Change at Week 16 | -9.3 | -3.2 | -11.2 | -5.9 |
Treatment with TALTZ resulted in improvement in dactylitis and enthesitis in patients with pre-existing dactylitis or enthesitis.
Treatment with TALTZ 80 mg Q4W resulted in an improvement in psoriatic skin lesions in patients with PsA.
Radiographic Response
Radiographic changes were assessed in PsA1. Inhibition of progression of structural damage was assessed radiographically and expressed as the change in modified total Sharp score (mTSS) at Week 16, compared to baseline. The total Sharp score was modified for psoriatic arthritis by addition of hand distal interphalangeal (DIP) joints.
TALTZ 80 mg Q4W inhibited the progression of structural joint damage (mTSS) compared to placebo at Week 16. The adjusted mean change from baseline in the mTSS was 0.13 for TALTZ 80 mg Q4W and 0.36 for placebo (difference in means TALTZ minus placebo: -0.23, 95% CI: (-0.42, -0.04)).
Physical Function
TALTZ treated patients showed improvement in physical function compared to patients treated with placebo as assessed by the Health Assessment Questionnaire-Disability Index (HAQ-DI) at Week 12 and 24. In both studies, the proportion of HAQ-DI responders (≥0.35 improvement in HAQ-DI score) was greater in the TALTZ 80 mg Q4W groups compared to placebo at week 12 and 24.
Other Health-Related Outcomes
General health status was assessed by the Short Form health survey (SF-36). At Week 12 in PsA1 and PsA2, patients treated with TALTZ showed greater improvement from baseline in the SF-36 physical component summary (PCS) score compared to patients treated with placebo but this improvement was not consistent in both studies for the SF-36 mental component summary (MCS) score. At Week 12, there was consistent evidence of effect in the physical-functioning, role-physical, bodily-pain, and general health domains but not in the social-functioning, role-emotional, vitality, and mental health domains.
Ankylosing Spondylitis
The safety and efficacy of TALTZ were assessed in 567 patients, in 2 randomized, double-blind, placebo-controlled studies (AS1 and AS2) in adult patients, age 18 years and older with active ankylosing spondylitis. Patients had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 despite non-steroidal anti-inflammatory drug (NSAID), corticosteroid, or disease modifying anti-rheumatic drug (DMARD) therapy. At baseline, patients had symptoms of AS for an average of 17 years across both studies. At baseline, approximately 32% of the patients were on a concomitant cDMARD. In AS2, all patients discontinued previous treatment with 1 or 2 TNF inhibitors due to either inadequate response or intolerance.
AS1 Study (NCT 02696785) evaluated 341 biologic-naive patients, who were treated with either TALTZ 80 mg or 160 mg at Week 0 followed by 80 mg every 2 weeks (Q2W) or 4 weeks (Q4W), adalimumab 40 mg every 2 weeks, or with placebo. Patients receiving placebo were re-randomized at Week 16 to receive TALTZ (160 mg starting dose, followed by 80 mg Q2W or Q4W). Patients receiving adalimumab were re-randomized at Week 16 to receive TALTZ (80 mg Q2W or Q4W). AS2 Study (NCT 02696798) evaluated 316 TNF-inhibitor experienced patients (90% were inadequate responders and 10% were intolerant to TNF inhibitors). All patients were treated with TALTZ 80 or 160 mg at Week 0 followed by 80 mg Q2W or Q4W, or with placebo. Patients receiving placebo were re-randomized at Week 16 to receive TALTZ (160 mg initial dose, followed by 80 mg Q2W or Q4W). The primary endpoint in both studies was the percentage of patients achieving an Assessment of Spondyloarthritis International Society 40 (ASAS40) response at Week 16.
Clinical Response
In both studies, patients treated with TALTZ 80 mg Q4W demonstrated greater improvements in ASAS40 and ASAS20 responses compared to placebo at Week 16 (Table 8 ). Responses were seen regardless of concomitant therapies. In AS2, responses were seen regardless of prior TNF-inhibitor exposure.
a Abbreviations: N = number of patients in the intent-to-treat population; NRI = Non-responder Imputation. | ||||||
b Patients with missing data were counted as non-responders. | ||||||
c At Week 0, patients received 80 mg or 160 mg of TALTZ. | ||||||
d An ASAS20 response is defined as a ≥20% improvement and an absolute improvement from baseline of ≥1 units (range 0 to 10) in ≥3 of 4 domains (Patient Global, Spinal Pain, Function, and Inflammation), and no worsening of ≥20% and ≥1 unit (range 0 to 10) in the remaining domain. An ASAS40 response is defined as a ≥40% improvement and an absolute improvement from baseline of ≥2 units in ≥3 of 4 domains without any worsening in the remaining domain. | ||||||
e Primary endpoint. | ||||||
| AS1 – biologic-naive | AS2 – TNF-inhibitor experienced | |||||
| TALTZ 80 mg Q4W c (N=81) | Placebo (N=87) | Difference from placebo (95% CI) | TALTZ 80 mg Q4W c (N=114) | Placebo (N=104) | Difference from placebo (95% CI) | |
| ASAS20 response d , % | 64 | 40 | 24 (9, 39) | 48 | 30 | 18 (6, 31) |
| ASAS40 response d,e , % | 48 | 18 | 30 (16, 43) | 25 | 13 | 13 (3, 23) |
The percent of patients achieving ASAS40 response by visit in AS1 is shown in Figure 2 .
Figure 2: ASAS40 Response through Week 16, NRI a

a Patients with missing data were counted as non-responders.
The improvement in the main components of the ASAS40 response criteria and other measures of disease activity are shown in Table 9 .
a Abbreviations: ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; BASMI = Bath Ankylosing Spondylitis Metrology Index; hsCRP = High sensitivity C-reactive protein. | ||||
b Mean changes are least-squares mean changes from baseline at Week 16. | ||||
c At Week 0, patients received 80 or 160 mg of TALTZ. | ||||
d Inflammation is the mean of patient-reported stiffness self-assessments (questions 5 and 6) in BASDAI. | ||||
| AS1 – biologic-naive | AS2 – TNF-inhibitor experienced | |||
| TALTZ 80 mg Q4W c (N=81) | Placebo (N=87) | TALTZ 80 mg Q4W c (N=114) | Placebo (N=104) | |
| ASAS Components | ||||
| Patient Global Assessment (0-10) | ||||
| Baseline | 6.9 | 7.1 | 8.0 | 7.8 |
| Mean Change from Baseline | -2.5 | -1.4 | -2.4 | -0.7 |
| Total Spinal Pain (0-10) | ||||
| Baseline | 7.2 | 7.4 | 7.9 | 7.8 |
| Mean Change from Baseline | -3.2 | -1.7 | -2.4 | -1.0 |
| BASFI (0-10) | ||||
| Baseline | 6.1 | 6.4 | 7.4 | 7.0 |
| Mean Change from Baseline | -2.4 | -1.2 | -1.7 | -0.6 |
| Inflammation (0-10) d | ||||
| Baseline | 6.5 | 6.8 | 7.2 | 7.2 |
| Mean Change from Baseline | -3.2 | -1.3 | -2.4 | -0.7 |
| Other Measures of Disease Activity | ||||
| BASDAI Score | ||||
| Baseline | 6.8 | 6.8 | 7.5 | 7.3 |
| Mean Change from Baseline | -2.9 | -1.4 | -2.2 | -0.9 |
| BASMI | ||||
| Baseline | 3.9 | 4.5 | 4.7 | 4.9 |
| Mean Change from Baseline | -0.5 | -0.1 | -0.3 | 0.0 |
| hsCRP (mg/L) | ||||
| Baseline | 12.2 | 16.0 | 20.2 | 16.0 |
| Mean Change from Baseline | -5.2 | 1.4 | -11.1 | 9.7 |
Health-Related Outcomes
General health status and quality of life was assessed by the Short Form health survey (SF-36). At Week 16, in AS1 and AS2, compared to placebo, patients treated with TALTZ showed greater improvement from baseline in the SF-36 physical component summary (PCS) score and the physical functioning, role physical, bodily pain, vitality, and general health domains, with no consistent improvements in the mental component summary (MCS), social functioning, role emotional, and mental health domains.
Non-radiographic Axial Spondyloarthritis
The efficacy and safety of TALTZ were assessed in a randomized, double-blind, 52-week placebo-controlled study (nr-axSpA1) (NCT 02757352) in patients ≥18 years of age with active axial spondyloarthritis for at least 3 months. Patients must have had objective signs of inflammation indicated by elevated C-reactive protein (CRP) (defined as greater than 5 mg/L), and/or sacroiliitis on magnetic resonance imaging (MRI), and no definitive radiographic evidence of structural damage on sacroiliac joints. Patients had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, and spinal pain ≥4 on a 0 to 10 Numerical Rating Scale (NRS). Patients must have been intolerant to or had an inadequate response to at least two NSAIDs. Patients were treated with either placebo or TALTZ 80 mg or 160 mg at Week 0, followed by either 80 mg every 2 weeks (Q2W) or 80 mg every 4 weeks (Q4W). Initiation and/or dose adjustment of concomitant medications (NSAIDs, cDMARDs, corticosteroids, analgesics) were permitted starting at Week 16. Patients were allowed to transition to use of open-label TALTZ 80 mg Q2W starting at Week 16 up to Week 44 at the discretion of the investigator.
At baseline, patients had symptoms of nr-axSpA for an average of 11 years. Approximately 39% of the patients were on a concomitant cDMARD.
The primary endpoint was the percentage of patients achieving an Assessment of Spondyloarthritis International Society 40 (ASAS40) response at Week 52. The ASAS40 response was also evaluated at Week 16 as a major secondary endpoint.
Clinical Response
At both Weeks 16 and 52, a greater proportion of patients treated with TALTZ 80 mg Q4W had ASAS40 response compared to patients treated with placebo (Table 10 ). The components of ASAS response criteria and CRP are shown in Table 11 .
a Abbreviations: N = number of patients in the intent-to-treat population; NRI = Non-responder Imputation. | ||||||
b Patients who initiated open-label TALTZ 80 mg Q2W, or discontinued initially randomized treatment and remained in the study, or were missing Week 16 or Week 52 data were counted as non-responders. | ||||||
c Starting at Week 16 and up to Week 44, patients who were determined as inadequate responders by investigators were given the option to make changes in their background therapy and/or transition to open label TALTZ 80 mg Q2W. | ||||||
d At Week 0, patients received 80 mg or 160 mg of TALTZ. | ||||||
e An ASAS40 response is defined as a ≥40% improvement and an absolute improvement from baseline of ≥2 units in ≥3 of 4 domains without any worsening in the remaining domain. | ||||||
| Week 16 | Week 52 | |||||
| TALTZ 80 mg Q4W c,d (N=96) | Placebo (N=105) | Difference from placebo (95% CI) | TALTZ 80 mg Q4W c,d (N=96) | Placebo (N=105) | Difference from placebo (95% CI) | |
| ASAS40 response e , % | 35.4 | 19.0 | 16.4 (4.2, 28.5) | 30.2 | 13.3 | 16.9 (5.6, 28.1) |
The improvement in the main components of the ASAS40 response criteria and other measures of disease activity at Week 16 are shown in Table 11 .
a Abbreviations: BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; BASMI = Bath Ankylosing Spondylitis Metrology Index; hsCRP = High sensitivity C-reactive protein; NRI = Non-responder Imputation. | ||
b At Week 0, patients received 80 or 160 mg of TALTZ. | ||
c Mean changes are least-squares mean changes from baseline at Week 16 using a mixed model for repeated measures adjusting for treatment group, screening MRI/CRP classification, visit, continuous baseline, interaction of visit with treatment, interaction of continuous baseline with visit. | ||
d Inflammation is the mean of patient-reported stiffness self-assessments (questions 5 and 6) in BASDAI questionnaire. | ||
| TALTZ 80 mg Q4W b (N=96) | Placebo (N=105) | |
| ASAS Components | ||
| Patient Global Assessment (0-10) | ||
| Baseline | 7.1 | 7.4 |
| Mean Change from Baseline c | -2.3 | -1.3 |
| Total Spinal Pain (0-10) | ||
| Baseline | 7.3 | 7.4 |
| Mean Change from Baseline c | -2.4 | -1.5 |
| BASFI (0-10) | ||
| Baseline | 6.4 | 6.7 |
| Mean Change from Baseline c | -2.0 | -1.3 |
| Inflammation (0-10) d | ||
| Baseline | 6.8 | 7.0 |
| Mean Change from Baseline c | -2.5 | -1.5 |
| Other Measures of Disease Activity | ||
| BASDAI Score (0-10) | ||
| Baseline | 7.0 | 7.2 |
| Mean Change from Baseline c | -2.2 | -1.5 |
| BASMI (0-10) | ||
| Baseline | 3.2 | 3.2 |
| Mean Change from Baseline c | -0.4 | -0.2 |
| hsCRP (mg/L) | ||
| Baseline | 12.4 | 14.3 |
| Mean Change from Baseline c | -8.0 | -3.0 |
The percent of patients achieving ASAS40 responses by visit is shown in Figure 3 .
Figure 3: ASAS40 Responses through Week 16, NRI a

a Patients with missing data were counted as non-responders.
Health-Related Outcomes
General health status and quality of life was assessed by the Short Form health survey (SF-36). At Week 16, compared to placebo, nr-axSpA patients treated with TALTZ 80 mg Q4W showed greater improvement from baseline in the SF-36 physical component summary (PCS) score and physical functioning, bodily pain, vitality, and social functioning domains, with no consistent improvements in the mental component summary (MCS), role physical, general health, role emotional, and mental health domains.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
TALTZ injection is a sterile, preservative free, clear and colorless to slightly yellow solution available in:
- a single-dose prefilled autoinjector or a single-dose prefilled syringe to deliver 80 mg ixekizumab.
- a single-dose prefilled syringe to deliver 40 mg ixekizumab.
- a single-dose prefilled syringe to deliver 20 mg ixekizumab.
TALTZ is supplied as:
| Pack Size | NDC Code | |
| Autoinjector | ||
| Carton of 1 | 0002-1445-11 | |
| 80 mg single-dose | Carton of 2 | 0002-1445-27 |
| Carton of 3 | 0002-1445-09 | |
| Prefilled syringe | ||
| 80 mg single-dose | Carton of 1 | 0002-7724-11 |
| 40 mg single-dose | Carton of 1 | 0002-8905-11 |
| 20 mg single-dose | Carton of 1 | 0002-8900-11 |
Storage and Handling
TALTZ is sterile and preservative-free. Discard any unused portion.
- TALTZ must be protected from light until use.
- Store refrigerated at 2°C to 8°C (36°F to 46°F).
- If needed, patients/caregivers may store TALTZ at room temperature up to 30°C (86°F) for up to 5 days in the original carton to protect from light. Once TALTZ has been stored at room temperature, do not return to the refrigerator and discard, if unused, within 5 days.
- Record the date when TALTZ is first removed from the refrigerator in the spaces provided on the carton.
- For the 2 or 3 autoinjector pack, remove a single autoinjector at a time leaving the remaining autoinjector(s) in the original carton in the refrigerator. Ensure the unrefrigerated TALTZ is protected from light.
- Do not freeze. Do not use TALTZ if it has been frozen.
- Do not shake.
- Discard the TALTZ single-dose autoinjector or syringe after use in a puncture-resistant container.
- Not made with natural rubber latex.
TALTZ 80 mg Autoinjector
| INSTRUCTIONS FOR USE |
| TALTZ ® [tol-t-s] (ixekizumab) injection, for subcutaneous use Autoinjector |
| This Instructions for Use contains information on how to inject TALTZ. |
 |
| Important Information You Need to Know Before Injecting TALTZ |
|
|
|
|
|
|
| INSTRUCTIONS FOR USE Before you use the TALTZ autoinjector, read and carefully follow all the step-by-step instructions. | ||
| Parts of the TALTZ autoinjector | ||
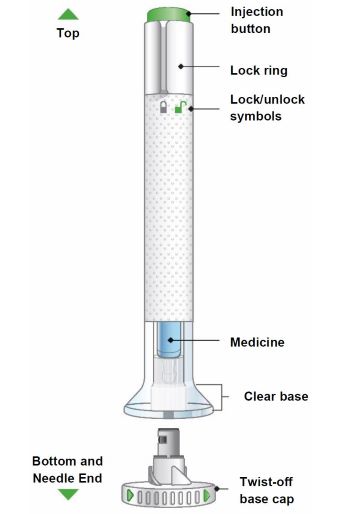 | ||
| Step 1 | Preparing to Inject TALTZ | |
| Step 1a |
|  |
| Step 1b | Gather the supplies needed for your injection:
| |
| Step 1c | 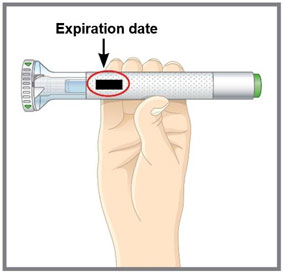 Figure A Figure A | Inspect the autoinjector.
|
| Step 1d | Wash your hands with soap and water before you inject TALTZ. | |
| Step 1e | 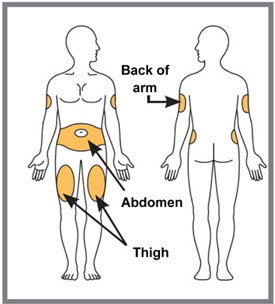 Figure B Figure B | Choose your injection site. You may inject in your stomach area (abdomen) or in your thigh, or in the back of your arm (see Figure B ). To inject in your arm, you will need someone to help you. Do not give an injection into areas where the skin is tender, bruised, red or hard, or in an area of skin that is affected by psoriasis. Do not inject within 1 inch of the navel (belly button). Alternate your injection sites.
|
| Step 1f | Prepare your skin. Clean your injection site with an alcohol wipe. Let the injection site dry before you inject TALTZ. | |
| Step 2 | Injecting TALTZ | |||
| Step 2a | 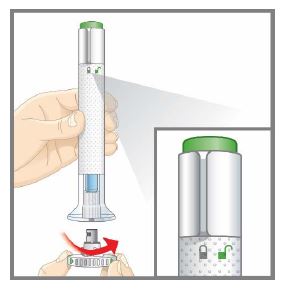 Figure C Figure C | Make sure the lock ring is in the lock position.
| ||
| Step 2b | 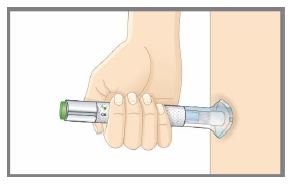 Figure D Figure D | Place the clear base flat and firmly against your skin at the injection site (see Figure D ). | ||
| Step 2c | 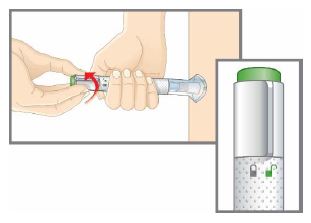 Figure E Figure E | While holding the clear base against your skin, turn the lock ring to the unlock position (see Figure E ). You are now ready to inject. | ||
| Step 2d | 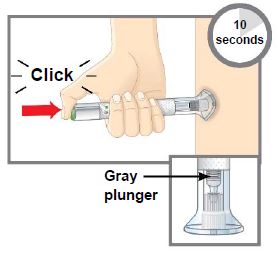 Figure F Figure F | Press the green injection button. There will be a loud click (see Figure F ).
| ||
| Step 3 | Disposing of TALTZ | |||
| Step 3a | 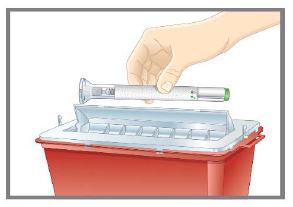 Figure G Figure G | |||
| ||||
| ||||
| ||||
| ||||
| Commonly asked questions and answers. | |
| Q. | What if I see bubbles in the TALTZ autoinjector? |
| A. | It is normal to have air bubbles in the autoinjector. |
| Q. | What if there is a drop of liquid on the tip of the needle when I remove the base cap? |
| A. | It is okay to see a drop of liquid on the tip of the needle. |
| Q. | What if I unlocked the autoinjector and pressed the green injection button before I twisted off the base cap? |
| A. | Do not remove the base cap. Dispose of the autoinjector and get a new one. |
| Q. | Do I need to hold the injection button down until the injection is complete? |
| A. | You do not need to hold the injection button down, but it may help you keep the autoinjector steady and firm against your skin. |
| Q. | What if the needle did not retract after my injection or I am not sure that the autoinjector worked in the right way? |
| A. | Do not touch the needle or replace the base cap. Store the autoinjector in a safe place (e.g., a household container as described in “Disposing of TALTZ” ) to avoid an accidental needlestick and contact Lilly (1-800-545-5979) for instructions on how to return the autoinjector. |
| Q. | What if I hear more than 2 clicks during my injection? Did I get my complete dose? |
| A. | You may hear a soft click right before the second loud click. This is the normal operation of the autoinjector. Do not remove the autoinjector from your skin until you hear the second loud click. |
| Q. | How can I tell if my injection is complete? |
| A. | After you press the green injection button, you will hear 2 loud clicks. The second click tells you that your injection is complete. You will also see the gray plunger at the top of the clear base. |
| Q. | What if the autoinjector is left at room temperature for longer than 30 minutes? |
| A. | If needed, the autoinjector may be left out of the refrigerator at room temperature up to 86°F (30°C) for up to 5 days if protected from light. Throw away TALTZ if not used within the 5-day period at room temperature. See " Storing TALTZ " for more detail. |
| If you have more questions about how to use the TALTZ autoinjector: | |
| |

| Storing TALTZ |
|
|
|
|
| Keep TALTZ and all medicines out of the reach of children. |
| Read the full Prescribing Information and Medication Guide for TALTZ inside this box to learn more about your medicine. |
|
| Eli Lilly and Company Indianapolis, IN 46285, USA US License Number 1891 TALTZ is a trademark of Eli Lilly and Company. Copyright © 2016, 2023, Eli Lilly and Company. All rights reserved. The TALTZ autoinjector meets the current dose accuracy and functional requirements of ISO 11608-1 and 11608-5. This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: December 2023 TALAI-0008-IFU-20231218 |
Mechanism of Action
Ixekizumab is a humanized IgG4 monoclonal antibody that selectively binds with the interleukin 17A (IL-17A) cytokine and inhibits its interaction with the IL-17 receptor. IL-17A is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Ixekizumab inhibits the release of proinflammatory cytokines and chemokines.