Get your patient on Cabergoline - Cabergoline tablet (Cabergoline)
Cabergoline - Cabergoline tablet prescribing information
INDICATIONS AND USAGE
Cabergoline tablets are indicated for the treatment of hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas.
DOSAGE AND ADMINISTRATION
The recommended dosage of cabergoline tablets for initiation of therapy is 0.25 mg twice a week. Dosage may be increased by 0.25 mg twice weekly up to a dosage of 1 mg twice a week according to the patient's serum prolactin level. Before initiating treatment, cardiovascular evaluation should be performed and echocardiography should be considered to assess for valvular disease.
Dosage increases should not occur more rapidly than every 4 weeks, so that the physician can assess the patient's response to each dosage level. If the patient does not respond adequately, and no additional benefit is observed with higher doses, the lowest dose that achieved maximal response should be used and other therapeutic approaches considered. Patients receiving long term treatment with cabergoline should undergo periodic assessment of their cardiac status and echocardiography should be considered.
After a normal serum prolactin level has been maintained for 6 months, cabergoline may be discontinued, with periodic monitoring of the serum prolactin level to determine whether or when treatment with cabergoline should be reinstituted. The durability of efficacy beyond 24 months of therapy with cabergoline has not been established.
CONTRAINDICATIONS
Cabergoline tablets are contraindicated in patients with:
- Uncontrolled hypertension or known hypersensitivity to ergot derivatives.
- History of cardiac valvular disorders, as suggested by anatomical evidence of valvulopathy of any valve, determined by pre-treatment evaluation including echocardiographic demonstration of valve leaflet thickening, valve restriction, or mixed valve restriction-stenosis. (See WARNINGS )
- History of pulmonary, pericardial, or retroperitoneal fibrotic disorders. (See WARNINGS )
ADVERSE REACTIONS
The safety of cabergoline tablets has been evaluated in more than 900 patients with hyperprolactinemic disorders. Most adverse events were mild or moderate in severity.
In a 4-week, double-blind, placebo-controlled study, treatment consisted of placebo or cabergoline at fixed doses of 0.125, 0.5, 0.75, or 1.0 mg twice weekly. Doses were halved during the first week. Since a possible dose-related effect was observed for nausea only, the four cabergoline treatment groups have been combined. The incidence of the most common adverse events during the placebo-controlled study is presented in the following table.
Incidence of Reported Adverse Events During the 4-Week, Double-Blind, Placebo-Controlled Trial
| Adverse Event• | Cabergoline (n=168) 0.125 to 1 mg two times a week | Placebo (n=20) |
| Number (percent) | ||
| Gastrointestinal Nausea Constipation Abdominal pain Dyspepsia Vomiting | 45 (27) 16 (10) 9 (5) 4 (2) 4 (2) | 4 (20) 0 1 (5) 0 0 |
| Central and Peripheral Nervous System Headache Dizziness Paresthesia Vertigo | 43 (26) 25 (15) 2 (1) 2 (1) | 5 (25) 1 (5) 0 0 |
| Body as a Whole Asthenia Fatigue Hot flashes | 15 (9) 12 (7) 2 (1) | 2 (10) 0 1 (5) |
| Psychiatric Somnolence Depression Nervousness | 9 (5) 5 (3) 4 (2) | 1 (5) 1 (5) 0 |
| Autonomic Nervous System Postural hypotension | 6 (4) | 0 |
| Reproductive – Female Breast pain Dysmenorrhea | 2 (1) 2 (1) | 0 0 |
| Vision Abnormal vision | 2 (1) | 0 |
•Reported at ≥1% for cabergoline
In the 8-week, double-blind period of the comparative trial with bromocriptine, cabergoline (at a dose of 0.5 mg twice weekly) was discontinued because of an adverse event in 4 of 221 patients (2%) while bromocriptine (at a dose of 2.5 mg two times a day) was discontinued in 14 of 231 patients (6%). The most common reasons for discontinuation from cabergoline were headache, nausea and vomiting (3, 2 and 2 patients respectively); the most common reasons for discontinuation from bromocriptine were nausea, vomiting, headache, and dizziness or vertigo (10, 3, 3, and 3 patients respectively). The incidence of the most common adverse events during the double-blind portion of the comparative trial with bromocriptine is presented in the following table.
Incidence of Reported Adverse Events During the 8-Week, Double-Blind Period of the Comparative Trial With Bromocriptine
| Adverse Event• | Cabergoline (n=221) | Bromocriptine (n=231) |
| Number (percent) | ||
| Gastrointestinal Nausea Constipation Abdominal pain Dyspepsia Vomiting Dry mouth Diarrhea Flatulence Throat irritation Toothache | 63 (29) 15 (7) 12 (5) 11 (5) 9 (4) 5 (2) 4 (2) 4 (2) 2 (1) 2 (1) | 100 (43) 21 (9) 19 (8) 16 (7) 16 (7) 2 (1) 7 (3) 3 (1) 0 0 |
| Central and Peripheral Nervous System Headache Dizziness Vertigo Paresthesia | 58 (26) 38 (17) 9 (4) 5 (2) | 62 (27) 42 (18) 10 (4) 6 (3) |
| Body as a Whole Asthenia Fatigue Syncope Influenza-like symptoms Malaise Periorbital edema Peripheral edema | 13 (6) 10 (5) 3 (1) 2 (1) 2 (1) 2 (1) 2 (1) | 15 (6) 18 (8) 3 (1) 0 0 2 (1) 1 |
| Psychiatric Depression Somnolence Anorexia Anxiety Insomnia Impaired concentration Nervousness | 7 (3) 5 (2) 3 (1) 3 (1) 3 (1) 2 (1) 2 (1) | 5 (2) 5 (2) 3 (1) 3 (1) 2 (1) 1 5 (2) |
| Cardiovascular Hot flashes Hypotension Dependent edema Palpitation | 6 (3) 3 (1) 2 (1) 2 (1) | 3 (1) 4 (2) 1 5 (2) |
| Reproductive – Female Breast pain Dysmenorrhea | 5 (2) 2 (1) | 8 (3) 1 |
| Skin and Appendages Acne Pruritus | 3 (1) 2 (1) | 0 1 |
| Musculoskeletal Pain Arthralgia | 4 (2) 2 (1) | 6 (3) 0 |
| Respiratory Rhinitis | 2 (1) | 9 (4) |
| Vision Abnormal vision | 2 (1) | 2 (1) |
•Reported at ≥1% for cabergoline
Other adverse events that were reported at an incidence of <1.0% in the overall clinical studies follow.
Body as a Whole: facial edema, influenza-like symptoms, malaise
Cardiovascular System: hypotension, syncope, palpitations
Digestive System: dry mouth, flatulence, diarrhea, anorexia
Metabolic and Nutritional System: weight loss, weight gain
Nervous System: somnolence, nervousness, paresthesia, insomnia, anxiety
Respiratory System: nasal stuffiness, epistaxis
Skin and Appendages: acne, pruritus
Special Senses: abnormal vision
Urogenital System: dysmenorrhea, increased libido
The safety of cabergoline has been evaluated in approximately 1,200 patients with Parkinson's disease in controlled and uncontrolled studies at dosages of up to 11.5 mg/day which greatly exceeds the maximum recommended dosage of cabergoline for hyperprolactinemic disorders. In addition to the adverse events that occurred in the patients with hyperprolactinemic disorders, the most common adverse events in patients with Parkinson's disease were dyskinesia, hallucinations, confusion, and peripheral edema. Heart failure, pleural effusion, pulmonary fibrosis, and gastric or duodenal ulcer occurred rarely. One case of constrictive pericarditis has been reported.
Postmarketing Surveillance data: The following events have been reported in association with cabergoline: cardiac valvulopathy and extracardiac fibrotic reactions, (See WARNINGS , Cardiac Valvulopathy and Extracardiac Fibrotic Reactions) .
Other events have been reported in association with cabergoline: impulse control/compulsive behavior symptoms, including hypersexuality, increased libido and pathological gambling (See PRECAUTIONS , Psychiatric ). In addition, cases of alopecia, aggression and psychotic disorder have been reported in patients taking cabergoline. Some of these reports have been in patients who have had prior adverse reactions to dopamine agonist products.
To report SUSPECTED ADVERSE REACTIONS, contact Somerset Therapeutics, LLC at 1-800-417-9175 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
DESCRIPTION
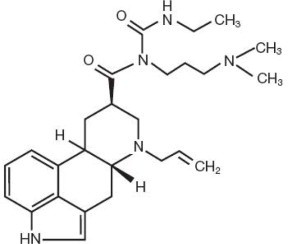
Cabergoline Tablets, USP contain cabergoline, USP a dopamine receptor agonist. The chemical name for cabergoline is 1-[(6-allylergolin-8β-yl)-carbonyl]-1-[3-(dimethylamino)propyl]-3-ethylurea. Its empirical formula is C 26 H 37 N 5 O 2 , and its molecular weight is 451.62. The structural formula is as follows:
Cabergoline, USP is a white powder soluble in ethyl alcohol, chloroform, and N, N-dimethylformamide (DMF); slightly soluble in 0.1N hydrochloric acid; very slightly soluble in n-hexane; and insoluble in water.
Cabergoline Tablets, USP for oral administration, contain 0.5 mg of cabergoline, USP. Inactive ingredients consist of citric acid anhydrous, croscarmellose sodium, magnesium stearate and microcrystalline cellulose.
CLINICAL PHARMACOLOGY
Mechanism of Action: The secretion of prolactin by the anterior pituitary is mainly under hypothalamic inhibitory control, likely exerted through release of dopamine by tuberoinfundibular neurons. Cabergoline is a long-acting dopamine receptor agonist with a high affinity for D 2 receptors. Results of in-vitro studies demonstrate that cabergoline exerts a direct inhibitory effect on the secretion of prolactin by rat pituitary lactotrophs. Cabergoline decreased serum prolactin levels in reserpinized rats. Receptor-binding studies indicate that cabergoline has low affinity for dopamine D 1 , α 1 - and α 2 -adrenergic, and 5-HT 1 - and 5-HT 2 -serotonin receptors.
Clinical Studies: The prolactin-lowering efficacy of cabergoline was demonstrated in hyperprolactinemic women in two randomized, double-blind, comparative studies, one with placebo and the other with bromocriptine. In the placebo-controlled study (placebo n=20; cabergoline n=168), cabergoline produced a dose-related decrease in serum prolactin levels with prolactin normalized after 4 weeks of treatment in 29%, 76%, 74% and 95% of the patients receiving 0.125, 0.5, 0.75, and 1.0 mg twice weekly respectively.
In the 8-week, double-blind period of the comparative trial with bromocriptine (cabergoline n=223; bromocriptine n=236 in the intent-to-treat analysis), prolactin was normalized in 77% of the patients treated with cabergoline at 0.5 mg twice weekly compared with 59% of those treated with bromocriptine at 2.5 mg twice daily. Restoration of menses occurred in 77% of the women treated with cabergoline, compared with 70% of those treated with bromocriptine. Among patients with galactorrhea, this symptom disappeared in 73% of those treated with cabergoline compared with 56% of those treated with bromocriptine.
Pharmacokinetics
Absorption: Following single oral doses of 0.5 mg to 1.5 mg given to 12 healthy adult volunteers, mean peak plasma levels of 30 to 70 picograms (pg)/mL of cabergoline were observed within 2 to 3 hours. Over the 0.5-to-7 mg dose range, cabergoline plasma levels appeared to be dose-proportional in 12 healthy adult volunteers and nine adult parkinsonian patients. A repeat-dose study in 12 healthy volunteers suggests that steady-state levels following a once-weekly dosing schedule are expected to be twofold to threefold higher than after a single dose. The absolute bioavailability of cabergoline is unknown. A significant fraction of the administered dose undergoes a first-pass effect. The elimination half-life of cabergoline estimated from urinary data of 12 healthy subjects ranged between 63 to 69 hours. The prolonged prolactin-lowering effect of cabergoline may be related to its slow elimination and long half-life.
Distribution: In animals, based on total radioactivity, cabergoline (and/or its metabolites) has shown extensive tissue distribution. Radioactivity in the pituitary exceeded that in plasma by >100-fold and was eliminated with a half-life of approximately 60 hours. This finding is consistent with the long-lasting prolactin-lowering effect of the drug. Whole body autoradiography studies in pregnant rats showed no fetal uptake but high levels in the uterine wall. Significant radioactivity (parent plus metabolites) detected in the milk of lactating rats suggests a potential for exposure to nursing infants. The drug is extensively distributed throughout the body. Cabergoline is moderately bound (40% to 42%) to human plasma proteins in a concentration-independent manner. Concomitant dosing of highly protein-bound drugs is unlikely to affect its disposition.
Metabolism: In both animals and humans, cabergoline is extensively metabolized, predominately via hydrolysis of the acylurea bond or the urea moiety. Cytochrome P-450 mediated metabolism appears to be minimal. Cabergoline does not cause enzyme induction and/or inhibition in the rat. Hydrolysis of the acylurea or urea moiety abolishes the prolactin-lowering effect of cabergoline, and major metabolites identified thus far do not contribute to the therapeutic effect.
Excretion: After oral dosing of radioactive cabergoline to five healthy volunteers, approximately 22% and 60% of the dose was excreted within 20 days in the urine and feces, respectively. Less than 4% of the dose was excreted unchanged in the urine. Nonrenal and renal clearances for cabergoline are about 3.2 L/min and 0.08 L/min, respectively. Urinary excretion in hyperprolactinemic patients was similar.
Special Populations
Renal Insufficiency: The pharmacokinetics of cabergoline were not altered in 12 patients with moderate-to-severe renal insufficiency as assessed by creatinine clearance.
Hepatic Insufficiency: In 12 patients with mild-to-moderate hepatic dysfunction (Child-Pugh score ≤10), no effect on mean cabergoline C max or area under the plasma concentration curve (AUC) was observed. However, patients with severe insufficiency (Child-Pugh score >10) show a substantial increase in the mean cabergoline C max and AUC, and thus necessitate caution.
Elderly: Effect of age on the pharmacokinetics of cabergoline has not been studied.
Food-Drug Interaction
In 12 healthy adult volunteers, food did not alter cabergoline kinetics.
Pharmacodynamics
Dose response with inhibition of plasma prolactin, onset of maximal effect, and duration of effect has been documented following single cabergoline doses to healthy volunteers (0.05 to 1.5 mg) and hyperprolactinemic patients (0.3 to 1 mg). In volunteers, prolactin inhibition was evident at doses >0.2 mg, while doses ≥0.5 mg caused maximal suppression in most subjects. Higher doses produce prolactin suppression in a greater proportion of subjects and with an earlier onset and longer duration of action. In 12 healthy volunteers, 0.5, 1, and 1.5 mg doses resulted in complete prolactin inhibition, with a maximum effect within 3 hours in 92% to 100% of subjects after the 1 and 1.5 mg doses compared with 50% of subjects after the 0.5 mg dose.
In hyperprolactinemic patients (N=51), the maximal prolactin decrease after a 0.6 mg single dose of cabergoline was comparable to 2.5 mg bromocriptine; however, the duration of effect was markedly longer (14 days vs. 24 hours). The time to maximal effect was shorter for bromocriptine than cabergoline (6 hours vs. 48 hours).
In 72 healthy volunteers, single or multiple doses (up to 2 mg) of cabergoline resulted in selective inhibition of prolactin with no apparent effect on other anterior pituitary hormones (GH, FSH, LH, ACTH, and TSH) or cortisol.
HOW SUPPLIED
Cabergoline Tablets, USP are white to off-white, scored, oval-shaped, flat face, beveled edge tablet, debossed with the letter "c" and the number "05" on either side of the break line and plain on the other side.
Cabergoline Tablets, USP are available as follows:
Bottles of 8 tablets NDC 70069-824-08