Get your patient on Rexulti (Brexpiprazole)
Rexulti patient education
Patient toolkit
Dosage & administration

Rexulti prescribing information
WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS and SUICIDAL THOUGHTS AND BEHAVIORS
WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS and SUICIDAL THOUGHTS AND BEHAVIORS
See full prescribing information for complete boxed warning.
- Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at increased risk of death. REXULTI is not approved for the treatment of patients with dementia-related psychosis without agitation associated with dementia due to Alzheimer's disease. (5.1 )
- Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients. Closely monitor all antidepressant-treated patients for clinical worsening and emergence of suicidal thoughts and behaviors. Safety and effectiveness of REXULTI have not been established in pediatric patients with MDD. (5.2 , 8.4 )
Increased Mortality in Elderly Patients with Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. REXULTI is not approved for the treatment of patients with dementia-related psychosis without agitation associated with dementia due to Alzheimer's disease [see Warnings and Precautions (5.1) ] .
Suicidal Thoughts and Behaviors
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric patients and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors [see Warnings and Precautions (5.2) ] . The safety and effectiveness of REXULTI have not been established in pediatric patients with MDD [see Warnings and Precautions (5.2) , Use in Specific Populations (8.4) ] .
| Warnings and Precautions (5.6 ) | 4/2026 |
INDICATIONS AND USAGE
REXULTI is indicated for:
- Adjunctive treatment to antidepressants for major depressive disorder (MDD) in adults
- Treatment of schizophrenia in adults and pediatric patients ages 13 years and older
- Treatment of agitation associated with dementia due to Alzheimer's disease
Limitations of Use:
REXULTI is not indicated as an as needed ("prn") treatment for agitation associated with dementia due to Alzheimer's disease [see Clinical Studies (14.3) ] .
DOSAGE AND ADMINISTRATION
Administer REXULTI orally once daily with or without food. (2 , 12.3 )
| Indication | Starting Dosage | Recommended Target Dosage | Maximum Dosage |
|---|---|---|---|
| MDD Adults (2.2 ) | 0.5 mg/day or 1 mg/day | 2 mg/day | 3 mg/day |
| Schizophrenia Adults (2.3 ) | 1 mg/day | 2 to 4 mg/day | 4 mg/day |
| Schizophrenia Pediatric (13 - 17 years) (2.3 ) | 0.5 mg/day | 2 to 4 mg/day | 4 mg/day |
| Agitation associated with dementia due to Alzheimer's disease (2.4 ) | 0.5 mg/day | 2 mg/day | 3 mg/day |
- Moderate to Severe Hepatic Impairment: Maximum recommended dosage is 2 mg once daily for patients with MDD or agitation associated with dementia due to Alzheimer's disease and 3 mg once daily for patients with schizophrenia. (2.5 )
- CrCl<60 mL/minute: Maximum recommended dosage is 2 mg once daily for patients with MDD or agitation associated with dementia due to Alzheimer's disease and 3 mg once daily for patients with schizophrenia. (2.6 )
- See Full Prescribing Information for dosage modifications for CYP2D6 poor metabolizers and for concomitant use with CYP inhibitors or inducers. (2.7 )
Administration Information
Administer REXULTI orally, once daily with or without food [see Clinical Pharmacology (12.3) ].
Recommended Dosage for Adjunctive Treatment of Major Depressive Disorder (Adults)
The recommended starting REXULTI dosage for the adjunctive treatment of MDD in adults is 0.5 mg or 1 mg orally once daily . Titrate to 1 mg once daily, then titrate to the target dosage of 2 mg once daily (based on the patient's clinical response and tolerability, increase the dosage at weekly intervals). The maximum recommended daily dosage is 3 mg. Periodically reassess to determine the continued need and appropriate dosage for treatment.
Recommended Dosage for Schizophrenia (Adults and Pediatric Patients 13 to 17 Years)
Adults
The recommended starting REXULTI dosage for the treatment of schizophrenia in adults is 1 mg orally once daily on Days 1 to 4. Titrate to 2 mg once daily on Day 5 through Day 7. On Day 8, the dosage can be increased to the maximum recommended daily dosage of 4 mg based on clinical response and tolerability. The recommended target dosage is 2 mg to 4 mg once daily.
Pediatric Patients (13 to 17 years of age)
The recommended starting REXULTI dosage for the treatment of schizophrenia in pediatric patients 13 to 17 years of age is 0.5 mg orally once daily on Days 1 to 4. On Days 5 through 7, titrate to 1 mg per day and on Day 8 titrate to 2 mg based on clinical response and tolerability. Weekly dose increases can be made in 1 mg increments. A recommended target dosage is 2 to 4 mg once daily. The maximum recommended daily dosage is 4 mg.
Recommended Dosage for Agitation Associated with Dementia Due to Alzheimer's Disease
The recommended starting REXULTI dosage for the treatment of agitation associated with dementia due to Alzheimer's disease is 0.5 mg orally once daily on Days 1 to 7 . Increase the dosage on Days 8 through 14 to 1 mg once daily, and on Day 15 to 2 mg once daily. The recommended target dose is 2 mg once daily. The dosage can be increased to the maximum recommended daily dosage of 3 mg once daily after at least 14 days, based on clinical response and tolerability.
Recommended Dosage in Patients with Hepatic Impairment
The maximum recommended dosage in patients with moderate to severe hepatic impairment (Child-Pugh score ≥7) is [see Use in Specific Populations (8.7) , Clinical Pharmacology (12.3) ]:
2 mg orally once daily in patients with MDD or agitation associated with dementia due to Alzheimer's disease, and
3 mg orally once daily in patients with schizophrenia
Recommended Dosage in Patients with Renal Impairment
The maximum recommended dosage in patients with creatinine clearance CrCl<60 mL/minute is [see Use in Specific Populations (8.8) , Clinical Pharmacology (12.3) ]:
2 mg orally once daily in patients with MDD or agitation associated with dementia due to Alzheimer's disease and
3 mg orally once daily in patients with schizophrenia
Dosage Modifications for CYP2D6 Poor Metabolizers and for Concomitant Use with CYP Inhibitors or Inducers
Dosage modifications are recommended in patients who are known cytochrome P450 (CYP) 2D6 poor metabolizers and in patients taking concomitant CYP3A4 inhibitors, CYP2D6 inhibitors, or strong CYP3A4 inducers (see Table 1 ). If the concomitant drug is discontinued, adjust the REXULTI dosage to its original level. If the concomitant CYP3A4 inducer is discontinued, reduce the REXULTI dosage to the original level over 1 to 2 weeks [see Drug Interactions (7.1) , Clinical Pharmacology (12.3) ] .
| Factors | Adjusted REXULTI Dosage |
|---|---|
| CYP2D6 Poor Metabolizers | |
| CYP2D6 poor metabolizers | Administer half of the recommended dosage. |
| Known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors | Administer a quarter of the recommended dosage. |
| Patients Taking CYP2D6 Inhibitors and/or CYP3A4 Inhibitors | |
| Strong CYP2D6 inhibitors In the clinical studies examining the use of REXULTI for the adjunctive treatment of MDD, dosage was not adjusted for strong CYP2D6 inhibitors (e.g., paroxetine, fluoxetine). Thus, CYP considerations are already factored into general dosing recommendations, and REXULTI may be administered without dosage adjustment in patients with MDD. | Administer half of the recommended dosage. |
| Strong CYP3A4 inhibitors | Administer half of the recommended dosage. |
| Strong/moderate CYP2D6 inhibitors with strong/moderate CYP3A4 inhibitors | Administer a quarter of the recommended dosage. |
| Patients Taking CYP3A4 Inducers | |
| Strong CYP3A4 inducers | Double the recommended dosage over 1 to 2 weeks. |
DOSAGE FORMS AND STRENGTHS
REXULTI tablets are available in 6 strengths:
- 0.25 mg tablets are light brown, round, shallow convex, bevel-edged body with "BRX" and "0.25" imprinted on one side.
- 0.5 mg tablets: are light orange, round, shallow convex, bevel-edged body with "BRX" and "0.5" imprinted on one side.
- 1 mg tablets are light yellow, round, shallow convex, bevel-edged body with "BRX" and "1" imprinted on one side.
- 2 mg tablets are light green, round, shallow convex, bevel-edged body with "BRX" and "2" imprinted on one side.
- 3 mg tablets are light purple, round, shallow convex, bevel-edged body with "BRX" and "3" imprinted on one side.
- 4 mg tablets are white, round, shallow convex, bevel-edged body with "BRX" and "4" imprinted on one side.
USE IN SPECIFIC POPULATIONS
Pregnancy: May cause extrapyramidal and/or withdrawal symptoms in neonates with third trimester exposure (8.1 )
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to REXULTI during pregnancy. For more information contact the National Pregnancy Registry for Psychiatric Medications at 1-866-961-2388 or visit http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/ .
Risk Summary
Adequate and well-controlled studies have not been conducted with REXULTI in pregnant women to inform drug-associated risks. However, neonates whose mothers are exposed to antipsychotic drugs, like REXULTI, during the third trimester of pregnancy are at risk for extrapyramidal and/or withdrawal symptoms. In animal reproduction studies, no teratogenicity was observed with oral administration of brexpiprazole to pregnant rats and rabbits during organogenesis at doses up to 73 and 146 times, respectively, of maximum recommended human dose (MRHD) of 4 mg/day on a mg/m 2 basis. However, when pregnant rats were administered brexpiprazole during the period of organogenesis through lactation, the number of perinatal deaths of pups was increased at 73 times the MRHD [see Data ] . The background risk of major birth defects and miscarriage for the indicated population(s) is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Extrapyramidal and/or withdrawal symptoms, including agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress and feeding disorder, have been reported in neonates whose mothers were exposed to antipsychotic drugs during the third trimester of pregnancy. These symptoms have varied in severity. Some neonates recovered within hours or days without specific treatment; others required prolonged hospitalization. Monitor neonates for extrapyramidal and/or withdrawal symptoms and manage symptoms appropriately.
Data
Animal Data
Pregnant rats were treated with oral doses of 3, 10, and 30 mg/kg/day (7.3, 24, and 73 times the MRHD on a mg/m 2 basis) of brexpiprazole during the period of organogenesis. Brexpiprazole was not teratogenic and did not cause adverse developmental effects at doses up to 73 times the MRHD.
Pregnant rabbits were treated with oral doses of 10, 30, and 150 mg/kg/day (49, 146, and 730 times the MRHD) of brexpiprazole during the period of organogenesis. Brexpiprazole was not teratogenic and did not cause adverse developmental effects at doses up to 146 times the MRHD. Findings of decreased body weight, retarded ossification, and increased incidences of visceral and skeletal variations were observed in fetuses at 730 times the MRHD, a dose that induced maternal toxicity.
In a study in which pregnant rats were administered oral doses of 3, 10, and 30 mg/kg/day (7.3, 24, and 73 times the MRHD) during the period of organogenesis and through lactation, the number of live-born pups was decreased, and early postnatal deaths increased at a dose 73 times the MRHD. Impaired nursing by dams, and low birth weight and decreased body weight gain in pups were observed at 73 times, but not at 24 times, the MRHD.
Lactation
Risk Summary
Lactation studies have not been conducted to assess the presence of brexpiprazole in human milk, the effects of brexpiprazole on the breastfed infant, or the effects of brexpiprazole on milk production. Brexpiprazole is present in rat milk. The development and health benefits of breastfeeding should be considered along with the mother's clinical need for REXULTI and any potential adverse effects on the breastfed infant from REXULTI or from the underlying maternal condition.
Pediatric Use
Schizophrenia
The safety and effectiveness of REXULTI for treatment of schizophrenia have been established in pediatric patients 13 years of age and older. Use of REXULTI in this population is supported by evidence from adequate and well-controlled studies in adults and pediatric patients with schizophrenia, pharmacokinetic data from adults and pediatric patients, and safety data in pediatric patients 13 to 17 years of age [see Warnings and Precautions (5.6) , Adverse Reactions (6.1) , Clinical Pharmacology (12.3) , Clinical Studies (14.2) ] .
The safety and effectiveness of REXULTI for the treatment of schizophrenia have not been established in pediatric patients less than 13 years of age.
Major Depressive Disorder
The safety and effectiveness of REXULTI for treatment of major depressive disorder have not been established in pediatric patients. Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric patients [see Boxed Warning , Warnings and Precautions (5.2) ] .
Irritability Associated with Autism Spectrum Disorder
The safety and effectiveness of REXULTI for the treatment of irritability associated with autism spectrum disorder have not been established in pediatric patients. Effectiveness was not demonstrated, in an 8-week, double-blind, placebo-controlled, flexible-dose clinical study conducted in 119 REXULTI-treated pediatric patients 5 to 17 years of age with irritability associated with autism spectrum disorder diagnosed by the Diagnostic and Statistical Manual of Mental Disorders, 5 th Edition [DSM-5] criteria. In this study, somnolence (including sedation) occurred at a higher rate than reported in other REXULTI studies evaluating adults and elderly patients (16% in REXULTI-treated pediatric patients versus 5% for placebo). The mean increase in age-and-gender adjusted body weight z-score from baseline to last visit was 0.3 for REXULTI-treated patients versus 0.1 for placebo-treated patients. Increases in age-and-gender adjusted body weight z-score of at least 0.5 SD from baseline was higher in REXULTI-treated patients versus placebo (19% versus 5%).
Of the 119 patients from this study, 95 patients entered the open-label treatment study and received up to 26 weeks of daily treatment with brexpiprazole. During the open-label treatment period, 2% of patients discontinued due to weight increase. In patients previously treated with REXULTI for 8 weeks, the mean increase in weight from the open-label study baseline to last visit was 4.5 kg and 26% of patients had an increase in age-and-gender-adjusted body weight z-score of at least 0.5 SD from baseline.
Juvenile rats were administered oral doses of brexpiprazole of 3, 10, and 20 mg/kg/day once daily beginning from weaning (postnatal day 21) through adulthood (postnatal day 90), followed by a 4-week recovery (non-dosing) period. Results were similar to those observed in previous repeat‑dose toxicity studies in adolescent (8-week-old) rats. Mortality occurred at the high dose of 20 mg/kg/day, as well as delayed sexual maturation in males and decreased rearing and motor activity. There was no evidence of neurotoxicity or effects on fertility and reproductive function. Histopathologic changes in reproductive organs and mammary glands occurred at all doses, were related to the pharmacology of brexpiprazole and were comparable to those in adult rats. All findings were at least partially reversible. Juvenile dogs were administered oral doses of brexpiprazole of 1, 3, and 30 mg/kg/day once daily starting at 8 or 9 weeks of age for 26 weeks, followed by an 8-week recovery (non-dosing) period. Decreases in body weight, lethargy, changes in heart rate, and immature male sex organs were observed at 30 mg/kg/day. These findings were at least partially reversible.
Geriatric Use
Antipsychotic drugs increase the risk of death in elderly patients with dementia-related psychosis. REXULTI is not approved for the treatment of patients with dementia-related psychosis [see Boxed Warning , Warnings and Precautions (5.1) ] .
Adjunctive Treatment of Major Depressive Disorder (MDD) and Schizophrenia
Of the total number of REXULTI-treated patients in the clinical studies for the adjunctive therapy to antidepressants for MDD and for schizophrenia, 248 (3%) were 65 years of age and older (which included 45 (18%) patients who were 75 years of age and older). Clinical studies of REXULTI in these patients did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients. In general, dosage selection for the treatment of MDD or schizophrenia in a geriatric patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Agitation Associated with Dementia Due to Alzheimer's Disease
The total number of REXULTI-treated patients 65 years of age and older in the clinical studies for agitation associated with dementia due to Alzheimer's disease (Studies 6 and 7) was 448 (86%) including 170 (33%) patients 65 to 74 years of age, 228 (44%) patients 75 to 84 years of age, and 50 (10%) patients 85 years of age and older [see Clinical Studies (14.3) ] .
In clinical studies of REXULTI for the treatment of agitation associated with dementia due to Alzheimer's disease did not include sufficient numbers of younger adult patients to determine if patients 65 years of age and older respond differently than younger adult patients.
CYP2D6 Poor Metabolizers
Dosage adjustment is recommended in known CYP2D6 poor metabolizers because these patients have higher brexpiprazole concentrations than normal metabolizers of CYP2D6. Approximately 8% of Caucasians and 3 to 8% of Black/African Americans cannot metabolize CYP2D6 substrates and are classified as poor metabolizers [see Dosage and Administration (2.7) , Clinical Pharmacology (12.3) ] .
Hepatic Impairment
The maximum recommended dosage in patients with moderate to severe hepatic impairment (Child-Pugh score ≥7) is lower than those with mild hepatic impairment and those with normal hepatic function [see Dosage and Administration (2.4) ] . Patients with moderate to severe hepatic impairment generally had higher exposure to brexpiprazole than patients with normal hepatic function [see Clinical Pharmacology (12.3) ] . Greater exposure may increase the risk of REXULTI-associated adverse reactions .
Renal Impairment
The maximum recommended dosage in patients with CrCl<60 mL/minute is lower than those with mild renal impairment and those with normal renal function [see Dosage and Administration (2.6) ] . Patients with renal impairment had higher exposure to brexpiprazole than patients with normal renal function [see Clinical Pharmacology (12.3) ] . Greater exposure may increase the risk of REXULTI-associated adverse reactions.
Other Specific Populations
The recommended dosage for REXULTI is the same in males and females, in different racial groups, and in smokers and nonsmokers [see Clinical Pharmacology (12.3) ] .
CONTRAINDICATIONS
REXULTI is contraindicated in patients with a known hypersensitivity to brexpiprazole or any of its components. Reactions have included rash, facial swelling, urticaria, and anaphylaxis.
WARNINGS AND PRECAUTIONS
- Cerebrovascular Adverse Reactions in Elderly Patients with Dementia-Related Psychosis: Increased incidence of cerebrovascular adverse reactions (e.g., stroke, transient ischemic attack) (5.3 )
- Neuroleptic Malignant Syndrome: Manage with immediate discontinuation and close monitoring. (5.4 )
- Tardive Dyskinesia: Discontinue if clinically appropriate. (5.5 )
- Metabolic Changes: Monitor for hyperglycemia/diabetes mellitus, dyslipidemia, and weight gain. (5.6 )
- Pathological Gambling and Other Compulsive Behaviors: Consider dose reduction or discontinuation. (5.7 )
- Leukopenia, Neutropenia, and Agranulocytosis: Perform complete blood counts (CBC) in patients with pre-existing low white blood cell count (WBC) or history of leukopenia or neutropenia. Consider discontinuing REXULTI if a clinically significant decline in WBC occurs in absence of other causative factors. (5.8 )
- Orthostatic Hypotension and Syncope: Monitor heart rate and blood pressure and warn patients with known cardiovascular or cerebrovascular disease, and risk of dehydration or syncope. (5.9 )
- Seizures: Use cautiously in patients with a history of seizures or with conditions that lower the seizure threshold. (5.11 )
- Potential for Cognitive and Motor Impairment: Use caution when operating machinery. (5.14 )
Increased Mortality in Elderly Patients with Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of 17 placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in the drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group.
Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. REXULTI is not approved for the treatment of patients with dementia-related psychosis without agitation associated with dementia due to Alzheimer's disease [see Boxed Warning , Warnings and Precautions (5.3) ] .
Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults
In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and over 4,400 pediatric patients, the incidence of suicidal thoughts and behaviors in patients 24 years of age and younger was greater in antidepressant-treated patients than in placebo-treated patients. The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1,000 patients treated are provided in Table 2 .
No suicides occurred in any of the pediatric studies. There were suicides in the adult studies, but the number was not sufficient to reach any conclusion about antidepressant drug effect on suicide.
| Age Range (years) | Drug-Placebo Difference in Number of Patients with Suicidal Thoughts or Behaviors per 1,000 Patients Treated |
|---|---|
| Increases Compared to Placebo | |
| <18 | 14 additional patients |
| 18 to 24 | 5 additional patients |
| Decreases Compared to Placebo | |
| 25 to 64 | 1 fewer patient |
| ≥65 | 6 fewer patients |
It is unknown whether the risk of suicidal thoughts and behaviors in children, adolescents, and young adults extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance studies in adults with MDD that antidepressants delay the recurrence of depression.
Monitor all antidepressant-treated patients for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of drug therapy and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behavior and to alert the healthcare provider. Consider changing the therapeutic regimen, including possibly discontinuing REXULTI, in patients whose depression is persistently worse or who are experiencing emergent suicidal thoughts or behaviors.
Cerebrovascular Adverse Reactions Including Stroke in Elderly Patients with Dementia-Related Psychosis
In placebo-controlled trials in elderly patients with dementia, patients randomized to risperidone, aripiprazole, and olanzapine had a higher incidence of stroke and transient ischemic attack, including fatal stroke. REXULTI is not approved for the treatment of patients with dementia-related psychosis without agitation associated with dementia due to Alzheimer's disease [see Boxed Warning , Warnings and Precautions (5.1) ].
Neuroleptic Malignant Syndrome (NMS)
Neuroleptic Malignant Syndrome (NMS), a potentially fatal symptom complex, has been reported in association with administration of antipsychotic drugs, including REXULTI.
Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status, and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis and cardiac dysrhythmia). Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure.
If NMS is suspected, immediately discontinue REXULTI and provide intensive symptomatic treatment and monitoring.
Tardive Dyskinesia
Tardive dyskinesia, a syndrome consisting of potentially irreversible, involuntary, dyskinetic movements, may develop in patients treated with antipsychotic drugs. The risk appears to be highest among the elderly, especially elderly women, but it is impossible to predict which patients will develop the syndrome. Whether antipsychotic drugs differ in their potential to cause tardive dyskinesia is unknown.
The risk of tardive dyskinesia and the likelihood that it will become irreversible appear to increase as the duration of treatment and the cumulative dose increases. The syndrome can develop after relatively brief treatment periods, at low doses. It may also occur after discontinuation of treatment.
Tardive dyskinesia may remit, partially or completely, if antipsychotic treatment is discontinued. Antipsychotic treatment itself may suppress (or partially suppress) the signs and symptoms of the syndrome, possibly masking the underlying process. The effect that symptomatic suppression has upon the long-term course of tardive dyskinesia is unknown.
Given these considerations, REXULTI should be prescribed in a manner most likely to reduce the occurrence of tardive dyskinesia. Chronic antipsychotic treatment should generally be reserved for patients who suffer from a chronic illness that 1) is known to respond to antipsychotic drugs and 2) for whom alternative, equally effective, but potentially less harmful treatments are not available or appropriate. In patients who do require chronic treatment, use the lowest dose and the shortest duration of treatment needed to produce a satisfactory clinical response. Periodically reassess the need for continued treatment.
If signs and symptoms of tardive dyskinesia appear in a patient treated with REXULTI, drug discontinuation should be considered. However, some patients may require treatment with REXULTI despite the presence of the syndrome.
Metabolic Changes
Atypical antipsychotic drugs, including REXULTI, have caused metabolic changes including hyperglycemia, diabetes mellitus, dyslipidemia, and body weight gain. Although all of the drugs in the class to date have been shown to produce some metabolic changes, each drug has its own specific risk profile.
Hyperglycemia/Diabetes Mellitus
Hyperglycemia and diabetes mellitus, in some cases extreme and associated with diabetic ketoacidosis hyperosmolar coma or death, have been reported in patients treated with atypical antipsychotics. There have been reports of hyperglycemia in patients treated with REXULTI. Assess fasting plasma glucose before or soon after initiation of antipsychotic medication and monitor periodically during long-term treatment.
Adjunctive Treatment of Major Depressive Disorder
In the 6-week placebo-controlled, fixed-dose clinical studies in adult patients with MDD, the proportions of patients with shifts in fasting glucose from normal (<100 mg/dL) to high (≥126 mg/dL) and borderline (≥100 and <126 mg/dL) to high were similar in patients treated with REXULTI and placebo. In the long-term, open-label depression studies, 5% of adult patients with normal baseline fasting glucose experienced a shift to high while taking REXULTI plus an antidepressant (ADT); 25% of patients with borderline fasting glucose experienced shifts to high. Combined, 9% of patients with normal or borderline fasting glucose experienced shifts to high fasting glucose during the long-term depression studies.
Schizophrenia (Adults)
In the 6-week placebo-controlled, fixed-dose clinical studies in adult patients with schizophrenia, the proportions of patients with shifts in fasting glucose from normal (<100 mg/dL) to high (≥126 mg/dL) or borderline (≥100 and <126 mg/dL) to high were similar in patients treated with REXULTI and placebo. In the long-term, open-label schizophrenia studies, 8% of adult patients with normal baseline fasting glucose experienced a shift from normal to high while taking REXULTI; 17% of patients with borderline fasting glucose experienced shifts from borderline to high. Combined, 10% of patients with normal or borderline fasting glucose experienced shifts to high fasting glucose during the long-term schizophrenia studies.
Schizophrenia [Pediatric Patients (13 to 17 years of age)]
In a 6-week, placebo-controlled study in pediatric patients with schizophrenia, the proportion of patients with shifts in fasting glucose from normal (<100 mg/dL) to high (≥126 mg/dL) or borderline (≥100 and <126 mg/dL) to high were similar in patients treated with REXULTI and placebo. In this study, 1.1% of pediatric patients with normal baseline fasting glucose experienced a shift from normal (<100 mg/dL) to high (≥126 mg/dL) while taking REXULTI.
In the 2-year, open-label study in pediatric patients with schizophrenia, 2.5% of pediatric patients with normal baseline fasting glucose experienced a shift from normal (<100 mg/dL) to high (≥126 mg/dL) while taking REXULTI.
Agitation Associated with Dementia Due to Alzheimer's Disease
In the 12-week placebo-controlled, fixed-dose studies in patients (51 to 90 years of age) with agitation associated with dementia due to Alzheimer's disease, the proportions of patients with shifts in fasting glucose from normal (<100 mg/dL) to high (≥126 mg/dL) or impaired (≥100 and <126 mg/dL) to high were similar in patients treated with REXULTI (14%) and patients treated with placebo (16%).
Of the patients who were previously treated with REXULTI for 12-weeks and continued into a 12-week, active-treatment extension study, 15% of patients with normal baseline fasting glucose experienced a shift from normal (<100 mg/dL) to high (≥126 mg/dL) fasting glucose while taking REXULTI; 30% of patients with impaired fasting glucose experienced shifts from impaired fasting glucose (≥100 and <126 mg/dL) to high fasting glucose. Combined, 20% of patients with normal or impaired fasting glucose experienced shifts to high fasting glucose.
Dyslipidemia
Atypical antipsychotics cause adverse alterations in lipids. Before or soon after initiation of antipsychotic medication, obtain a fasting lipid profile at baseline and monitor periodically during treatment.
Adjunctive Treatment of Major Depressive Disorder
In the 6-week placebo-controlled, fixed-dose clinical studies in adult patients with MDD, changes in fasting total cholesterol, LDL cholesterol, and HDL cholesterol were similar in REXULTI- and placebo-treated patients. Table 3 shows the proportions of patients with changes in fasting triglycerides.
| Proportion of Patients with Shifts Baseline to Post-Baseline | ||||
|---|---|---|---|---|
| Triglycerides | Placebo | 1 mg/day | 2 mg/day | 3 mg/day |
| Normal to High (<150 mg/dL to ≥200 and <500 mg/dL) | 6% (15/257) denotes n/N where N=the total number of patients who had a measurement at baseline and at least one post-baseline result; n=the number of patients with shift | 5% (7/145) | 13% (15/115) | 9% (13/150) |
| Normal/Borderline to Very High (<200 mg/dL to ≥500 mg/dL) | 0% (0/309) | 0% (0/177) | 0.7% (1/143) | 0% (0/179) |
In the long-term, open-label depression studies, shifts in baseline fasting cholesterol from normal to high were reported in 9% (total cholesterol), 3% (LDL cholesterol), and shifts in baseline from normal to low were reported in 14% (HDL cholesterol) of patients taking REXULTI. Of patients with normal baseline triglycerides, 17% experienced shifts to high, and 0.2% experienced shifts to very high. Combined, 0.6% of patients with normal or borderline fasting triglycerides experienced shifts to very high fasting triglycerides during the long-term depression studies.
Schizophrenia (Adults)
In the 6-week placebo-controlled, fixed-dose clinical studies in adult patients with schizophrenia, changes in fasting total cholesterol, LDL cholesterol, and HDL cholesterol were similar in REXULTI- and placebo-treated patients. Table 4 shows the proportions of patients with changes in fasting triglycerides.
| Proportion of Patients with Shifts Baseline to Post-Baseline | ||||
|---|---|---|---|---|
| Triglycerides | Placebo | 1 mg/day | 2 mg/day | 4 mg/day |
| Normal to High (<150 mg/dL to ≥200 and <500 mg/dL) | 6% (15/253) denotes n/N where N=the total number of patients who had a measurement at baseline and at least one post-baseline result; n=the number of patients with shift | 10% (7/72) | 8% (19/232) | 10% (22/226) |
| Normal/Borderline to Very High (<200 mg/dL to ≥500 mg/dL) | 0% (0/303) | 0% (0/94) | 0% (0/283) | 0.4% (1/283) |
In the long-term, open-label schizophrenia studies in adult patients, shifts in baseline fasting cholesterol from normal to high were reported in 6% (total cholesterol), 2% (LDL cholesterol), and shifts in baseline from normal to low were reported in 17% (HDL cholesterol) of patients taking REXULTI. Of patients with normal baseline triglycerides, 13% experienced shifts to high, and 0.4% experienced shifts to very high triglycerides. Combined, 0.6% of patients with normal or borderline fasting triglycerides experienced shifts to very high fasting triglycerides during the long-term schizophrenia studies.
Schizophrenia [Pediatric Patients (13 to 17 years of age)]
The safety and efficacy of REXULTI have not been established in patients under the age of 13 years. In a 6-week, placebo-controlled study in pediatric patients with schizophrenia, no clinically meaningful changes in fasting cholesterol, LDL cholesterol, HDL cholesterol, and triglycerides were observed between the REXULTI and placebo groups.
In the 2-year, open-label study in pediatric patients with schizophrenia, shifts in baseline fasting total cholesterol from normal to high (<170 to ≥200 mg/dL) were reported in 9.6% of patients taking REXULTI, and shifts in baseline HDL cholesterol from normal to low (>45 to <40 mg/dL) were reported in 17.2% of patients taking REXULTI. Of patients with normal baseline triglycerides, 24.5% experienced shifts from normal to high (<90 to ≥130 mg/dL).
Agitation Associated with Dementia Due to Alzheimer's Disease
In the 12-week placebo-controlled, fixed-dose clinical studies in patients (55 to 90 years of age) with agitation associated with dementia due to Alzheimer's disease, changes in total cholesterol, LDL cholesterol, and HDL cholesterol were similar in REXULTI- and placebo-treated patients.
Table 5 shows the proportions of patients with changes in fasting triglycerides in REXULTI- and placebo-treated patients.
| Proportion of Patients with Shifts Baseline to Post-Baseline | ||||
|---|---|---|---|---|
| Triglycerides | Placebo | 1 mg/day | 2 mg/day | 3 mg/day |
| Normal to High (<150 and 200 to <500 mg/dL) | 6% (10/157) denotes n/N where N=the total number of patients who had a measurement at baseline and at least one post-baseline result; n=the number of patients with shift | 9% (9/99) | 13% (17/133) | 6% (6/94) |
| Borderline to High (150 and <200mg/dL to 200 and <500 mg/dL) | 12% (3/26) | 33% (2/6) | 28% (7/25) | 26% (6/23) |
| Normal/Borderline to High (<200 mg/dL to 200 and <500 mg/dL) | 7% (13/183) | 11% (11/105) | 15% (24/158) | 10% (12/117) |
Of the patients who were previously treated with REXULTI for 12 weeks and continued into a 12-week, active-treatment extension study, 9% of patients taking REXULTI showed shifts in baseline fasting total cholesterol from normal (<200 mg/dL) to high (≥240 mg/dL), and 16% of patients taking REXULTI showed shifts in baseline HDL cholesterol from normal to low (≥40 to <40 mg/dL). Of the patients with normal baseline triglycerides, 18% experienced shifts from normal (<150 mg/dL) to high (200 to <500 mg/dL).
Weight Gain
Weight gain has been observed in patients treated with atypical antipsychotics, including REXULTI. Monitor weight at baseline and frequently thereafter.
Adjunctive Treatment of Major Depressive Disorder: Table 6 shows weight gain data at last visit and percentage of adult patients with ≥7% increase in body weight at endpoint from the 6-week placebo-controlled, fixed-dose clinical studies in patients with MDD.
| Placebo | 1 mg/day | 2 mg/day | 3 mg/day | |
|---|---|---|---|---|
| n=407 | n=225 | n=187 | n=228 | |
| Mean Change from Baseline (kg) at Last Visit | ||||
| All Patients | +0.3 | +1.3 | +1.6 | +1.6 |
| Proportion of Patients with a ≥7% Increase in Body Weight (kg) at Any Visit (n/N N=the total number of patients who had a measurement at baseline and at least one post-baseline result; n=the number of patients with a shift ≥7% ) | ||||
| 2% | 5% | 5% | 2% | |
| (8/407) | (11/225) | (9/187) | (5/228) | |
In the long-term, open-label depression studies, 4% of patients discontinued due to weight increase. REXULTI was associated with mean change from baseline in weight of 2.9 kg at Week 26 and 3.1 kg at Week 52. In the long-term, open-label depression studies, 30% of patients demonstrated a ≥7% increase in body weight, and 4% demonstrated a ≥7% decrease in body weight.
Schizophrenia (Adults)
Table 7 shows weight gain data at last visit and percentage of adult patients with ≥7% increase in body weight at endpoint from the 6-week placebo-controlled, fixed-dose clinical studies in adult patients with schizophrenia.
| Placebo | 1 mg/day | 2 mg/day | 4 mg/day | |
|---|---|---|---|---|
| n=362 | n=120 | n=362 | n=362 | |
| Mean Change from Baseline (kg) at Last Visit | ||||
| All Patients | +0.2 | +1.0 | +1.2 | +1.2 |
| Proportion of Patients with a ≥7% Increase in Body Weight (kg) at Any Visit ( denotes n/N where N=the total number of patients who had a measurement at baseline and at least one post-baseline result; n=the number of patients with a shift ≥7% n/N) | ||||
| 4% | 10% | 11% | 10% | |
| (15/362) | (12/120) | (38/362) | (37/362) | |
In the long-term, open-label schizophrenia studies in adult patients, 0.6% of patients discontinued due to weight increase. REXULTI was associated with mean change from baseline in weight of 1.3 kg at Week 26 and 2.0 kg at Week 52. In the long-term, open label schizophrenia studies, 20% of patients demonstrated a ≥7% increase in body weight, and 10% demonstrated a ≥7% decrease in body weight.
Schizophrenia [Pediatric Patients (13 to 17 years of age)]
In a 6-week, placebo-controlled study in pediatric patients with schizophrenia, no patients discontinued due to weight increase. The mean increase in weight from baseline to last visit was 0.8 kg in the REXULTI group and no changes were seen in the placebo groups. The percentage of pediatric patients demonstrating a ≥7% increase in body weight was 8.2% in the REXULTI group and 4.9% in the placebo group.
In the 2-year, open label study in pediatric patients with schizophrenia, 0.3% of patients discontinued due to weight increase. The mean increase in weight from the open-label study baseline to last visit was 3.9 kg. To adjust for normal growth, z-scores were derived (measured in standard deviations [SD]), which normalize for natural growth of children and adolescents by comparisons to age- and gender- matched population standards. A z-score change <0.5 SD is considered not clinically significant. In this study, the mean change in z-score from open-label baseline to last visit was 0.00 SD for body weight, while 21.8% of patients had an increase in age-and-gender-adjusted body weight z-score of at least 0.5 SD from baseline. When treating pediatric patients, weight gain should be monitored and assessed against that expected for normal growth.
Agitation Associated with Dementia Due to Alzheimer's Disease
In the 12-week placebo-controlled, fixed-dose clinical studies in patients (51 to 90 years of age) with agitation associated with dementia due to Alzheimer's disease, the proportion of the patients with a ≥7% increase in body weight (kg) at any visit were 2% in REXULTI compared to 0% in placebo group.
In patients who were previously treated with REXULTI for 12 weeks and who continued into a 12-week, active-treatment extension study, there was no mean change in weight (kg) from baseline to last visit in association with REXULTI. In this extension study, 4% of patients demonstrated ≥7% increase in body weight, and 5% demonstrated a ≥7% decrease in body weight from baseline to last visit.
Pathological Gambling and Other Compulsive Behaviors
Post-marketing case reports suggest that patients can experience intense urges, particularly for gambling, and the inability to control these urges while taking REXULTI. Other compulsive urges, reported less frequently, include sexual urges, shopping, eating, or binge eating, and other impulsive or compulsive behaviors. Because patients may not recognize these behaviors as abnormal, it is important for prescribers to ask patients or their caregivers specifically about the development of new or intense gambling urges, compulsive sexual urges, compulsive shopping, binge or compulsive eating, or other urges while being treated with REXULTI. In some cases, although not all, urges were reported to have stopped when the dose was reduced, or the medication was discontinued. Compulsive behaviors may result in harm to the patient and others if not recognized. Consider dose reduction or stopping the medication if a patient develops such urges.
Leukopenia, Neutropenia, and Agranulocytosis
Leukopenia and neutropenia have been reported during treatment with antipsychotic agents. Agranulocytosis (including fatal cases) has been reported with other agents in this class.
Possible risk factors for leukopenia and neutropenia include pre-existing low white blood cell count (WBC) or absolute neutrophil count (ANC) and history of drug-induced leukopenia or neutropenia. In patients with a pre-existing low WBC or ANC or a history of drug-induced leukopenia or neutropenia, perform a complete blood count (CBC) frequently during the first few months of therapy. In such patients, consider discontinuation of REXULTI at the first sign of a clinically significant decline in WBC in the absence of other causative factors.
Monitor patients with clinically significant neutropenia for fever or other symptoms or signs of infection and treat promptly if such symptoms or signs occur. Discontinue REXULTI in patients with absolute neutrophil count <1000/mm 3 and follow their WBC until recovery.
Orthostatic Hypotension and Syncope
Atypical antipsychotics cause orthostatic hypotension and syncope. Generally, the risk is greatest during initial dose titration and when increasing the dose. In the short-term, placebo-controlled clinical studies of REXULTI plus ADT in adult patients with MDD, the incidence of orthostatic hypotension-related adverse reactions in REXULTI plus ADT-treated patients compared to placebo plus ADT-treated patients included: dizziness (2% versus 2%) and orthostatic hypotension (0.1% versus 0%). In the short-term, placebo-controlled clinical studies of REXULTI in adult patients with schizophrenia, the incidence of orthostatic hypotension-related adverse reactions in REXULTI-treated patients compared to placebo patients included: dizziness (2% versus 2%), orthostatic hypotension (0.4% versus 0.2%), and syncope (0.1% versus 0%). In 12-week, placebo-controlled clinical studies of REXULTI in patients with agitation associated with dementia due to Alzheimer's disease, the incidence of orthostatic hypotension-related adverse reactions in patients treated with REXULTI compared to patients treated with placebo included: dizziness (3% versus 3%), orthostatic hypotension (1% versus 1%), and syncope (0.2% versus 0.8%).
Orthostatic vital signs should be monitored in patients who are vulnerable to hypotension (e.g., elderly patients, patients with dehydration, hypovolemia, concomitant treatment with antihypertensive medication), patients with known cardiovascular disease (history of myocardial infarction, ischemic heart disease, heart failure, or conduction abnormalities), and patients with cerebrovascular disease. REXULTI has not been evaluated in patients with a recent history of myocardial infarction or unstable cardiovascular disease. Such patients were excluded from the premarketing clinical studies.
Falls
Antipsychotics, including REXULTI, may cause somnolence, postural hypotension, motor, and sensory instability, which may lead to falls and, consequently, fractures or other injuries. For patients with diseases, conditions, or medications that could exacerbate these effects, complete fall risk assessments when initiating antipsychotic treatment and recurrently for patients on long-term antipsychotic treatment.
Seizures
Like other antipsychotic drugs, REXULTI may cause seizures. This risk is greatest in patients with a history of seizures or with conditions that lower the seizure threshold. Conditions that lower the seizure threshold may be more prevalent in older patients.
Body Temperature Dysregulation
Atypical antipsychotics may disrupt the body's ability to reduce core body temperature. Strenuous exercise, exposure to extreme heat, dehydration, and anticholinergic medications may contribute to an elevation in core body temperature; use REXULTI with caution in patients who may experience these conditions.
Dysphagia
Esophageal dysmotility and aspiration have been associated with antipsychotic drug use. Antipsychotic drugs, including REXULTI, should be used cautiously in patients at risk for aspiration.
Potential for Cognitive and Motor Impairment
REXULTI, like other antipsychotics, may cause somnolence and has the potential to impair judgment, thinking, or motor skills. In the 6-week placebo-controlled clinical studies in patients with MDD, somnolence (including sedation and hypersomnia) was reported in 4% of REXULTI plus ADT-treated patients compared to 1% of placebo plus ADT-treated patients.
In the 6-week placebo-controlled clinical studies in adult patients with schizophrenia, somnolence (including sedation and hypersomnia) was reported in 5% of REXULTI-treated patients compared to 3% of placebo-treated patients.
In the 12-week placebo-controlled, fixed-dose clinical studies in patients (51 to 90 years of age) with agitation associated with dementia due to Alzheimer's disease, somnolence (including sedation) was reported in 3% of patients treated with REXULTI compared to 1% of patients treated with placebo.
Patients should be cautioned about operating hazardous machinery, including motor vehicles, until they are reasonably certain that REXULTI therapy does not affect them adversely.
ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Increased Mortality in Elderly Patients with Dementia-Related Psychosis [see Boxed Warning , Warnings and Precautions (5.1) ]
- Suicidal Thoughts and Behaviors in Adolescents and Young Adults [see Boxed Warning , Warnings and Precautions (5.2) ]
- Cerebrovascular Adverse Reactions Including Stroke in Elderly Patients with Dementia-Related Psychosis [see Warnings and Precautions (5.3) ]
- Neuroleptic Malignant Syndrome (NMS) [see Warnings and Precautions (5.4) ]
- Tardive Dyskinesia [see Warnings and Precautions (5.5) ]
- Metabolic Changes [see Warnings and Precautions (5.6) ]
- Pathological Gambling and Other Compulsive Behaviors [see Warnings and Precautions (5.7) ]
- Leukopenia, Neutropenia, and Agranulocytosis [see Warnings and Precautions (5.8) ]
- Orthostatic Hypotension and Syncope [see Warnings and Precautions (5.9) ]
- Falls [see Warnings and Precautions (5.10) ]
- Seizures [see Warnings and Precautions (5.11) ]
- Body Temperature Dysregulation [see Warnings and Precautions (5.12) ]
- Dysphagia [see Warnings and Precautions (5.13) ]
- Potential for Cognitive and Motor Impairment [see Warnings and Precautions (5.14) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The most common adverse reactions in adult patients in clinical trials (≥5%) were weight increased, akathisia, headache, somnolence, and insomnia.
The most common adverse reactions in pediatric patients in clinical trials (≥5%) were weight increased, somnolence, headache, akathisia, and nasopharyngitis.
Brexpiprazole has been evaluated for safety in 12,550 adult patients who participated in multiple-dose clinical trials for major depressive disorder, schizophrenia, agitation associated with dementia due to Alzheimer's disease, attention deficit hyperactivity disorder (ADHD), post-traumatic stress disorder (PTSD), bipolar mania, and borderline personality disorder (BPD). Among them, 3,870 patients were treated with brexpiprazole for at least 180 days, and 1,910 patients were treated for at least one year of exposure.
Additionally, brexpiprazole has been evaluated for safety in 119 pediatric patients who participated in short-term trials, and 314 patients in long-term multiple-dose clinical trials for pediatric schizophrenia and autism spectrum disorders (ASD).
Adjunctive Treatment in Major Depressive Disorder (MDD)
The safety of REXULTI was evaluated in 1054 adult patients (18 to 65 years of age) diagnosed with MDD who participated in two 6-week placebo-controlled, fixed-dose clinical studies in patients with major depressive disorder in which REXULTI was administered at doses of 1 mg to 3 mg daily as adjunctive treatment to continued antidepressant therapy; patients in the placebo group continued to receive antidepressant therapy [see Clinical Studies (14.1) ] .
Adverse Reactions Reported as Reasons for Discontinuation of Treatment
A total of 3% (17/643) of REXULTI-treated patients and 1% (3/411) of placebo-treated patients discontinued due to adverse reactions.
Adverse Reactions in REXULTI Studies for Adjunctive MDD in Adults
Adverse reactions associated with the adjunctive use of REXULTI (incidence of 2% or greater and adjunctive REXULTI incidence greater than adjunctive placebo) that occurred during acute therapy (up to 6-weeks in patients with MDD) are shown in Table 8.
| Placebo (N=411) % | REXULTI | ||||
|---|---|---|---|---|---|
| 1 mg/day (N=226) % | 2 mg/day (N=188) % | 3 mg/day (N=229) % | All REXULTI (N=643) % | ||
| Gastrointestinal Disorders | |||||
| Constipation | 1 | 3 | 2 | 1 | 2 |
| General Disorders and Administration Site Conditions | |||||
| Fatigue | 2 | 3 | 2 | 5 | 3 |
| Infections and Infestations | |||||
| Nasopharyngitis | 2 | 7 | 1 | 3 | 4 |
| Investigations | |||||
| Weight Increased | 2 | 7 | 8 | 6 | 7 |
| Blood Cortisol Decreased | 1 | 4 | 0 | 3 | 2 |
| Metabolism and Nutrition | |||||
| Increased Appetite | 2 | 3 | 3 | 2 | 3 |
| Nervous System Disorders | |||||
| Akathisia | 2 | 4 | 7 | 14 | 9 |
| Headache | 6 | 9 | 4 | 6 | 7 |
| Somnolence | 0.5 | 4 | 4 | 6 | 5 |
| Tremor | 2 | 4 | 2 | 5 | 4 |
| Dizziness | 1 | 1 | 5 | 2 | 3 |
| Psychiatric Disorders | |||||
| Anxiety | 1 | 2 | 4 | 4 | 3 |
| Restlessness | 0 | 2 | 3 | 4 | 3 |
Dose-Related Adverse Reactions in the Adjunctive MDD Studies
In Studies 1 and 2, among the adverse reactions that occurred at ≥2% incidence in the patients treated with REXULTI plus ADT, the incidences of akathisia and restlessness increased with increases in dose.
Schizophrenia
Adults
The safety of REXULTI was evaluated in 852 adult patients (18 to 65 years of age) diagnosed with schizophrenia who participated in two 6-week placebo-controlled, fixed-dose clinical studies in which REXULTI was administered at daily doses of 1 mg, 2 mg, and 4 mg [see Clinical Studies (14.2) ].
Adverse Reactions Occurring at an Incidence of 2% or More in Patients Treated with REXULTI for Schizophrenia
Adverse reactions associated with REXULTI (incidence of 2% or greater and REXULTI incidence greater than placebo) during short-term (up to 6 weeks) studies in adult patients with schizophrenia are shown in Table 9.
| Placebo (N=368) % | REXULTI | ||||
|---|---|---|---|---|---|
| 1 mg/day (N=120) % | 2 mg/day (N=368) % | 4 mg/day (N=364) % | ALL REXULTI (N=852) % | ||
| Gastrointestinal Disorders | |||||
| Dyspepsia | 2 | 6 | 2 | 3 | 3 |
| Diarrhea | 2 | 1 | 3 | 3 | 3 |
| Investigations | |||||
| Weight Increased | 2 | 3 | 4 | 4 | 4 |
| Blood Creatine Phosphokinase Increased | 1 | 4 | 2 | 2 | 2 |
| Nervous System Disorders | |||||
| Akathisia | 5 | 4 | 5 | 7 | 6 |
| Tremor | 1 | 2 | 2 | 3 | 3 |
| Sedation | 1 | 2 | 2 | 3 | 2 |
Pediatric Patients (13 to 17 years of age)]
The safety of REXULTI was evaluated in 110 pediatric patients (13 to 17 years of age) diagnosed with schizophrenia who participated in a 6-week, placebo-controlled, clinical study in which REXULTI was administered at daily doses of 2 mg to 4 mg [see Clinical Studies (14.2) ].
Adverse Reactions Occurring at an Incidence of 2% or More in Pediatric Patients (13 to 17 years of age) Treated with REXULTI for Schizophrenia
Adverse reactions associated with REXULTI (incidence of 2% or greater and REXULTI incidence greater than placebo) during short-term (up to 6 weeks) study in pediatric patients with schizophrenia are shown in Table 10.
| Placebo (N=104) % | REXULTI (N=110) % | |
|---|---|---|
| Gastrointestinal Disorders | ||
| Nausea | 4 | 6 |
| Nervous System Disorders | ||
| Akathisia | 3 | 4 |
| Extrapyramidal Symptoms Extrapyramidal Symptoms includes: blepharospasm, dystonia, extrapyramidal disorder, eye movement disorder, hypokinesia, muscle rigidity, musculoskeletal stiffness, psychomotor hyperactivity, tremor | 3 | 6 |
| Headache | 5 | 6 |
Agitation Associated with Dementia Due to Alzheimer's Disease
The safety of REXULTI was evaluated in 503 patients (51 to 90 years of age), with a probable diagnosis of agitation associated with dementia due to Alzheimer's disease, who participated in two 12-week placebo-controlled, fixed-dose clinical studies in which REXULTI was administered at daily doses of 2 mg to 3 mg [see Clinical Studies (14.3) ].
Discontinuation of Treatment Due to Adverse Reactions
In two 12-week placebo-controlled, fixed-dose, clinical studies, a total of 5.6% (28/503) of patients treated with REXULTI and 4.8% (12/251) of patients treated with placebo discontinued due to adverse reactions.
Adverse Reactions Occurring at an Incidence of 2% or More in Patients Treated with REXULTI for Agitation Associated with Dementia Due to Alzheimer's Disease
Adverse reactions associated with REXULTI (incidence ≥2% and greater than placebo) during the 12-week fixed-dose clinical studies in geriatric patients for treatment of agitation associated with dementia due to Alzheimer's disease are shown in Table 11.
| Placebo (N=251) % | REXULTI | ||||
|---|---|---|---|---|---|
| 1 mg/day 1 mg once day REXULTI dosage is not a recommended dosage for the treatment of agitation associated with dementia due to Alzheimer's disease [see Dosage and Administration (2.4) ] . (N=137) % | 2 mg/day (N=213) % | 3 mg/day (N=153) % | ALL REXULTI (N=503) % | ||
| Infections and Infestations | |||||
| Nasopharyngitis | 2 | 4 | 2 | 3 | 3 |
| Urinary Tract Infection | 1 | 2 | 3 | 3 | 3 |
| Nervous System Disorders | |||||
| Dizziness Dizziness and vertigo are grouped to Dizziness | 2 | 1 | 5 | 3 | 3 |
| Headache | 8 | 9 | 9 | 7 | 8 |
| Somnolence Sedation and somnolence are grouped to Somnolence. | 1 | 2 | 3 | 4 | 3 |
| Psychiatric Disorders | |||||
| Insomnia Initial insomnia and insomnia are grouped to Insomnia | 3 | 5 | 5 | 2 | 4 |
Extrapyramidal Symptoms
Adjunctive Treatment of Major Depressive Disorder
The incidence of reported extrapyramidal symptoms (EPS)-related adverse reactions, excluding akathisia, was 6% for REXULTI plus ADT-treated patients versus 3% for placebo plus ADT-treated patients. The incidence of akathisia events for REXULTI plus ADT-treated patients was 9% versus 2% for placebo plus ADT-treated patients.
In the 6-week placebo-controlled MDD studies, data was objectively collected on the Simpson-Angus Rating Scale (SAS) for EPS, the Barnes Akathisia Rating Scale (BARS) for akathisia and the Abnormal Involuntary Movement Score (AIMS) for dyskinesia. The mean change from baseline at last visit for REXULTI plus ADT-treated patients for the SAS, BARS and AIMS was comparable to placebo-treated patients. The percentage of patients who shifted from normal to abnormal was greater in REXULTI plus ADT-treated patients versus placebo plus ADT-treated patients for the BARS (4% versus 0.6%) and the SAS (4% versus 3%).
Schizophrenia (Adults)
The incidence of reported EPS-related adverse reactions, excluding akathisia, was 5% for REXULTI-treated adult patients versus 4% for placebo-treated patients. The incidence of akathisia events for REXULTI-treated adult patients was 6% versus 5% for placebo-treated patients.
In the 6-week placebo-controlled, fixed-dose schizophrenia studies in adults, data was objectively collected on the Simpson-Angus Rating Scale (SAS) for EPS, the Barnes Akathisia Rating Scale (BARS) for akathisia and the Abnormal Involuntary Movement Scale (AIMS) for dyskinesia. The mean change from baseline at last visit for REXULTI-treated patients for the SAS, BARS and AIMS was comparable to placebo-treated patients. The percentage of patients who shifted from normal to abnormal was greater in REXULTI-treated patients versus placebo for the BARS (2% versus 1%) and the SAS (7% versus 5%).
Schizophrenia [Pediatric Patients (13 to 17 years of age)]
The incidence of reported EPS-related adverse reactions, excluding akathisia, was 6.4% for REXULTI-treated pediatric patients versus 2.9% for placebo-treated patients. The incidence of akathisia events for REXULTI-treated pediatric patients was 3.6% versus 2.9% for placebo-treated patients.
In the 6-week placebo-and active controlled, schizophrenia study in pediatric patients, data was objectively collected on the Simpson-Angus Rating Scale (SAS) for EPS, the Barnes Akathisia Rating Scale (BARS) for akathisia and the Abnormal Involuntary Movement Scale (AIMS) for dyskinesia. The mean change from baseline at last visit for REXULTI-treated pediatric patients for the SAS, BARS and AIMS was comparable to placebo-treated patients. The percentage of patients who shifted from normal to abnormal was greater in REXULTI-treated patients versus placebo for the BARS (0.9% versus 0%) and the SAS (5.5% versus 2.9%).
Agitation Associated with Dementia Due to Alzheimer's Disease
The incidence of reported EPS-related adverse reactions, excluding akathisia, was 3% for REXULTI-treated patients versus 2% for placebo-treated patients. The incidence of akathisia events for REXULTI-treated patients was 1% versus 0% for placebo-treated patients.
In the 12-week placebo-controlled, fixed-dose studies in agitation associated with dementia due to Alzheimer's disease, data was objectively collected on the Simpson-Angus Rating Scale (SAS) for EPS, the Barnes Akathisia Rating Scale (BARS) for akathisia and the Abnormal Involuntary Movement Scale (AIMS) for dyskinesia. The mean change from baseline at last visit for REXULTI-treated patients for the SAS, BARS and AIMS was comparable to placebo-treated patients. The percentage of patients who shifted from normal to abnormal was greater in REXULTI-treated patients versus placebo for the SAS (6% versus 2%).
Dystonia
Symptoms of dystonia may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. While these symptoms can occur at low doses, they occur more frequently and with greater severity with high potency and at higher doses of first-generation antipsychotic drugs. An elevated risk of acute dystonia is observed in males and younger age groups.
Other Adverse Reactions Observed during Clinical Trial Evaluation of REXULTI
Other adverse reactions (≥1% frequency and greater than placebo) within the short-term, placebo-controlled trials in adult patients with MDD and schizophrenia are shown below. The following listing does not include adverse reactions: 1) already listed in previous tables or elsewhere in the labeling, 2) for which a drug cause was remote, 3) which were so general as to be uninformative, 4) which were not considered to have clinically significant implications, or 5) which occurred at a rate equal to or less than placebo.
Eye Disorders: Vision Blurred
Gastrointestinal Disorders: Nausea, Dry Mouth, Salivary Hypersecretion, Abdominal Pain, Flatulence
Investigations: Blood Prolactin Increased
Musculoskeletal and Connective Tissue Disorders: Myalgia
Psychiatric Disorders: Abnormal Dreams
Skin and Subcutaneous Tissue Disorders: Hyperhidrosis
Pediatric Patients (13 to 17 years of age)
In a short-term, randomized, double-blind, placebo- controlled study in pediatric patients 13 to 17 years of age with schizophrenia, safety was assessed in 110 patients in which 100 received REXULTI for at least 6 weeks. In a 2 year, open-label study in pediatric patients 13 to 17 years of age with schizophrenia, safety was assessed in 294 patients of which 251 received REXULTI for at least 6 months and 216 for at least 12 months. The total number of patients who completed the study was 178, with 50 patients treated for at least 2 years. Adverse reactions reported in clinical studies for this age group were generally similar to those observed in adult patients.
Hyperprolactinemia
In a 6-week, placebo-controlled study in pediatric patients with schizophrenia, a 3.3 ng/mL mean increase (from baseline to last visit) was observed in the REXULTI group (versus a 2.8 ng/mL mean decrease in the placebo group) in females. Additionally, more female subjects in the REXULTI group (28.9%, n=13) compared to the placebo group (4.7%, n=2) had shifts from normal (≤30 ng/mL) prolactin levels at baseline to abnormal (>30 ng/mL) during the course of treatment. In males, overall mean shifts in the REXULTI group were not consistent with an increase in prolactin however, more male subjects in the REXULTI group (21.4%, n=9) compared to the placebo group (7.0%, n=3) had shifts from normal (≤20 ng/mL) prolactin levels at baseline to abnormal (>20 ng/mL) during the course of treatment. One subject in the study experienced TEAE of galactorrhea without elevated prolactin.
In a 2-year open label study, the observed mean increase (from baseline to last visit) in the REXULTI group was 1.93 ng/mL in females and 3.13 ng/mL in males.
Postmarketing Experience
The following adverse reaction has been identified during post-approval use of REXULTI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Nervous System disorders: Neuroleptic Malignant Syndrome
DRUG INTERACTIONS
| Factors | Dosage Adjustments for REXULTI (2.7 ) |
| Strong CYP2D6 REXULTI may be administered without dosage adjustment in patients with MDD when administered with strong CYP2D6 inhibitors (e.g., paroxetine, fluoxetine). or CYP3A4 inhibitors | Administer half of recommended dosage. |
| Strong/moderate CYP2D6 with Strong/moderate CYP3A4 inhibitors | Administer a quarter of the recommended dosage. |
| Known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors | Administer a quarter of the recommended dosage. |
| Strong CYP3A4 inducers | Double the recommended dosage and further adjust based on clinical response. |
Drugs Having Clinically Important Interactions with REXULTI
See Table 12 for clinically important drug interactions with REXULTI.
| Strong CYP3A4 Inhibitors | |
| Clinical Impact: | Concomitant use of REXULTI with strong CYP3A4 inhibitors increased the exposure of brexpiprazole compared to the use of REXULTI alone [see Clinical Pharmacology (12.3) ] . |
| Intervention: | With concomitant use of REXULTI with a strong CYP3A4 inhibitor, reduce the REXULTI dosage [see Dosage and Administration (2.7) ] . |
| Strong CYP2D6 Inhibitors In the clinical studies examining the adjunctive use of REXULTI in the treatment of MDD, dosage was not adjusted for strong CYP2D6 inhibitors (e.g., paroxetine, fluoxetine). Thus, CYP considerations are already factored into general dosing recommendations, and REXULTI may be administered without dosage adjustment in patients with MDD. | |
| Clinical Impact: | Concomitant use of REXULTI with strong CYP2D6 inhibitors increased the exposure of brexpiprazole compared to the use of REXULTI alone [see Clinical Pharmacology (12.3) ] . |
| Intervention: | With concomitant use of REXULTI with a strong CYP2D6 inhibitor, reduce the REXULTI dosage [see Dosage and Administration (2.7) ] . |
| Both CYP3A4 Inhibitors and CYP2D6 Inhibitors | |
| Clinical Impact: | Concomitant use of REXULTI with 1) a strong CYP3A4 inhibitor and a strong CYP2D6 inhibitor; or 2) a moderate CYP3A4 inhibitor and a strong CYP2D6 inhibitor; or 3) a strong CYP3A4 inhibitor and a moderate CYP2D6 inhibitor; or 4) a moderate CYP3A4 inhibitor and a moderate CYP2D6 inhibitor increased the exposure of brexpiprazole compared to the use of REXULTI alone [see Clinical Pharmacology (12.3) ]. |
| Intervention: | With concomitant use of REXULTI with 1) a strong CYP3A4 inhibitor and a strong CYP2D6 inhibitor; or 2) a moderate CYP3A4 inhibitor and a strong CYP2D6 inhibitor; or 3) a strong CYP3A4 inhibitor and a moderate CYP2D6 inhibitor; or 4) a moderate CYP3A4 inhibitor and a moderate CYP2D6 inhibitor, decrease the REXULTI dosage [see Dosage and Administration (2.7) ]. |
| Strong CYP3A4 Inducers | |
| Clinical Impact: | Concomitant use of REXULTI and a strong CYP3A4 inducer decreased the exposure of brexpiprazole compared to the use of REXULTI alone [see Clinical Pharmacology (12.3) ]. |
| Intervention: | With concomitant use of REXULTI with a strong CYP3A4 inducer, increase the REXULTI dosage [see Dosage and Administration (2.7) ]. |
Drugs Having No Clinically Important Interactions with REXULTI
Based on pharmacokinetic studies, no dosage adjustment of REXULTI is required when administered concomitantly with CYP2B6 inhibitors (e.g., ticlopidine) or gastric pH modifiers (e.g., omeprazole). Additionally, no dosage adjustment for substrates of CYP2D6 (e.g., dextromethorphan), CYP3A4 (e.g., lovastatin), CYP2B6 (e.g., bupropion), BCRP (e.g., rosuvastatin), or P-gp (e.g., fexofenadine) is required when administered concomitantly with REXULTI.
DESCRIPTION
Brexpiprazole, an atypical antipsychotic, is available as REXULTI ® (brexpiprazole) tablets. Brexpiprazole is 7-{4-[4-(1-Benzothiophen-4-yl)piperazin-1-yl]butoxy}quinolin-2(1 H )-one. The empirical formula is C 25 H 27 N 3 O 2 S, and its molecular weight is 433.57. The chemical structure is:
 |
REXULTI tablets are for oral administration and are available in 0.25 mg, 0.5 mg, 1 mg, 2 mg, 3 mg, and 4 mg strengths. Inactive ingredients include lactose monohydrate, corn starch, microcrystalline cellulose, hydroxypropyl cellulose, low-substituted hydroxypropyl cellulose, magnesium stearate, hypromellose, and talc. Colorants include titanium dioxide, iron oxide, and ferrosoferric oxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
The mechanism of action of REXULTI in the adjunctive treatment of major depressive disorder, treatment of agitation associated with dementia due to Alzheimer's disease, or treatment of schizophrenia is unknown. However, the efficacy of REXULTI may be mediated through a combination of partial agonist activity at serotonin 5-HT 1A and dopamine D 2 receptors, and antagonist activity at serotonin 5-HT 2A receptors.
Pharmacodynamics
Brexpiprazole has affinity (expressed as K i ) for multiple monoaminergic receptors including serotonin 5-HT 1A (0.12 nM), 5-HT 2A (0.47 nM), 5-HT 2B (1.9 nM), 5-HT 7 (3.7 nM), dopamine D 2 (0.30 nM), D 3 (1.1 nM), and noradrenergic α 1A (3.8 nM), α 1B (0.17 nM), α 1D (2.6 nM), and α 2C (0.59 nM) receptors. Brexpiprazole acts as a partial agonist at the 5-HT 1A , D 2 , and D 3 receptors and as an antagonist at 5-HT 2A , 5-HT 2B , 5-HT 7 , α 1A , α 1B , α 1D , and α 2C receptors. Brexpiprazole also exhibits affinity for histamine H 1 receptor (19 nM) and for muscarinic M 1 receptor (67% inhibition at 10 µM).
Cardiac Electrophysiology
At a dose 3 times the MRHD for the treatment of schizophrenia and 4 times the MRHD for adjunctive therapy to antidepressants for the treatment of MDD or agitation associated with dementia due to Alzheimer's disease, REXULTI does not prolong the QTc interval to any clinically relevant extent.
Pharmacokinetics
Absorption
After single-dose administration of REXULTI tablets, the peak plasma brexpiprazole concentrations occurred within 4 hours after administration, and the absolute oral bioavailability was 95%. Brexpiprazole steady-state concentrations were attained within 10 to 12 days of dosing.
REXULTI can be administered with or without food. Administration of a 4 mg REXULTI tablet with a standard high-fat meal did not significantly affect the C max or AUC of brexpiprazole. After single and multiple once daily dose administration, brexpiprazole exposure (C max and AUC) increased in proportion to the dose administered. In vitro studies of brexpiprazole did not indicate that brexpiprazole is a substrate of efflux transporters such as MDRI (P-gp) and BCRP.
Distribution
The volume of distribution of brexpiprazole following intravenous administration is high (1.56 ± 0.42 L/kg), indicating extravascular distribution. Brexpiprazole is highly protein bound in plasma (greater than 99%) to serum albumin and α1-acid glycoprotein, and its protein binding is not affected by renal or hepatic impairment. Based on results of in vitro studies, brexpiprazole protein binding is not affected by warfarin, diazepam, or digitoxin.
Elimination
Metabolism
Based on in vitro metabolism studies of brexpiprazole using recombinant human cytochrome P450 (CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4), the metabolism of brexpiprazole was shown to be mainly mediated by CYP3A4 and CYP2D6.
In vivo brexpiprazole is metabolized primarily by CYP3A4 and CYP2D6 enzymes. After single- and multiple-dose administrations, brexpiprazole and its major metabolite, DM-3411, were the predominant drug moieties in the systemic circulation. At steady-state, DM-3411 represented 23% to 48% of brexpiprazole exposure (AUC) in plasma. DM-3411 is considered not to contribute to the therapeutic effects of brexpiprazole.
Based on in vitro data, brexpiprazole showed little to no inhibition of CYP450 isozymes.
Excretion
Following a single oral dose of [ 14 C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in the urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine, and approximately 14% of the oral dose was recovered unchanged in the feces. Apparent oral clearance of a brexpiprazole oral tablet after once daily administration is 19.8 (±11.4) mL/h/kg. After multiple once-daily administrations of REXULTI, the terminal elimination half-lives of brexpiprazole and its major metabolite, DM-3411, were 91 hours and 86 hours, respectively.
Studies in Specific Populations
Exposure of brexpiprazole in specific populations are summarized in Figure 1. Population pharmacokinetic (PK) analysis indicated exposure of brexpiprazole in patients with moderate renal impairment was higher compared to patients with normal renal function.
Figure 1 The Effect of Intrinsic Factors on Brexpiprazole Pharmacokinetics

Pediatric Patients
A multiple dose PK study (0.5, 1, 2, 3 or 4 mg/day) has been conducted in 43 pediatric patients aged 13 years to 17 years old. Population PK analysis indicated systemic exposure (C max and AUC) of brexpiprazole in pediatric patients (13 to 17 years of age) was comparable to that in adult patients across the dose range from 0.5 to 4 mg.
Drug Interaction Studies
Effect of other drugs on the exposures of brexpiprazole are summarized in Figure 2. Based on simulation, a 5.1-fold increase in AUC values at steady-state is expected when extensive metabolizers of CYP2D6 are administered with both strong CYP2D6 and CYP3A4 inhibitors. A 4.8-fold increase in mean AUC values at steady-state is expected in poor metabolizers of CYP2D6 administered with strong CYP3A4 inhibitors [see Drug Interactions (7.1) ].
Figure 2 The Effect of Other Drugs on Brexpiprazole Pharmacokinetics

The effect of REXULTI on the exposures of other drugs are summarized in Figure 3 .
Figure 3 The Effect of REXULTI on Pharmacokinetics of Other Drugs

NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lifetime carcinogenicity studies were conducted in ICR mice and Sprague Dawley rats. Brexpiprazole was administered orally for two years to male and female mice at doses of 0.75, 2, and 5 mg/kg/day (0.9 to 6.1 times the oral MRHD of 4 mg/day based on mg/m 2 body surface area) and to male and female rats at doses of 1, 3, and 10 mg/kg and 3, 10, and 30 mg/kg/day, respectively (2.4 to 24 and 7.3 to 73 times the oral MRHD, males and females). In female mice, the incidence of mammary gland adenocarcinoma was increased at all doses, and the incidence of adenosquamous carcinoma was increased at 2.4 and 6.1 times the MRHD. No increase in the incidence of tumors was observed in male mice. In the rat study, brexpiprazole was not carcinogenic in either sex at doses up to 73 times the MRHD.
Proliferative and/or neoplastic changes in the mammary and pituitary glands of rodents have been observed following chronic administration of antipsychotic drugs and are considered to be prolactin mediated. The potential for increasing serum prolactin level of brexpiprazole was shown in both mice and rats. The relevance for human risk of the findings of prolactin-mediated endocrine tumors in rodents is unknown.
Mutagenesis
Brexpiprazole was not mutagenic when tested in the in vitro bacterial reverse mutation assay (Ames test). Brexpiprazole was negative for clastogenic activity in the in vivo micronucleus assay in rats and was not genotoxic in the in vivo/in vitro unscheduled DNA synthesis assay in rats. In vitro with mammalian cells brexpiprazole was clastogenic but only at doses that induced cytotoxicity. Based on the weight of evidence, brexpiprazole is not considered to present a genotoxic risk to humans.
Impairment of Fertility
Female rats were treated with oral doses of 0.3, 3, or 30 mg/kg/day (0.7, 7.3, and 73 times the oral MRHD on a mg/m 2 basis) prior to mating with untreated males and continuing through conception and implantation. Estrus cycle irregularities and decreased fertility were observed at 3 and 30 mg/kg/day. Prolonged duration of pairing and increased preimplantation losses were observed at 30 mg/kg/day.
Male rats were treated with oral doses of 3, 10, or 100 mg/kg/day (7.3, 24, and 240 times the oral MRHD on a mg/m 2 basis) for 63 days prior to mating with untreated females and throughout the 14 days of mating. No differences were observed in the duration of mating or fertility indices in males at any dose of brexpiprazole.
CLINICAL STUDIES
Adjunctive Treatment of Major Depressive Disorder
The efficacy of REXULTI in the adjunctive treatment of major depressive disorder (MDD) was evaluated in two 6-week double-blind, placebo-controlled, fixed-dose studies of adult patients meeting DSM-IV-TR criteria for MDD, with or without symptoms of anxiety, who had an inadequate response to prior antidepressant therapy (1 to 3 courses) in the current episode and who had also demonstrated an inadequate response throughout the 8 weeks of prospective antidepressant treatment (with escitalopram, fluoxetine, paroxetine controlled-release, sertraline, duloxetine delayed release, or venlafaxine extended release). Inadequate response during the prospective antidepressant treatment phase was defined as having persistent symptoms without substantial improvement throughout the course of treatment.
Patients in Study 1 (NCT01360645) were randomized to REXULTI 2 mg once a day or placebo. Patients in Study 2 (NCT01360632) were randomized to REXULTI 1 or 3 mg once a day or placebo. For patients randomized to REXULTI, all patients initiated treatment at 0.5 mg once daily during Week 1. At Week 2, the REXULTI dosage was increased to 1 mg in all treatment groups, and either maintained at 1 mg or increased to 2 mg or 3 mg once daily, based on treatment assignment, from Week 3 onwards. The dosages were then maintained for the 4 remaining weeks.
The primary endpoint was change from baseline to Week 6 in the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item clinician-related scale used to assess the degree of depressive symptomatology, with 0 representing no symptoms and 60 representing worst symptoms.
At randomization, the mean MADRS total score was 27. In Studies 1 and 2, REXULTI (plus ADT) 2 mg once daily and 3 mg once daily were superior to placebo plus ADT in reducing mean MADRS total scores. Results from the primary efficacy parameters for both fixed dose studies are shown below in Table 13. Figure 4 below shows the time course of response based on the primary efficacy measure (MADRS) in Study 1.
| Study | Treatment Group | N | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference Difference (drug minus placebo) in least-squares mean change from baseline (95% CI) | |
|---|---|---|---|---|---|---|
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: unadjusted confidence interval | ||||||
| 1 | REXULTI (2 mg/day) + ADT Dosages statistically significantly superior to placebo | 175 | 26.9 (5.7) | -8.4 (0.6) | -3.2 (-4.9, -1.5) | |
| Placebo + ADT | 178 | 27.3 (5.6) | -5.2 (0.6) | -- | ||
| 2 | REXULTI (1 mg/day) + ADT | 211 | 26.5 (5.6) | -7.6 (0.5) | -1.3 (-2.7, 0.1) | |
| REXULTI (3 mg/day) + ADT | 213 | 26.5 (5.3) | -8.3 (0.5) | -2.0 (-3.4, -0.5) | ||
| Placebo + ADT | 203 | 26.5 (5.2) | -6.3 (0.5) | -- | ||
An examination of population subgroups did not suggest differential response based on age, gender, race, or choice of prospective antidepressant.
Figure 4 Change from Baseline in MADRS Total Score by Study Visit (Week) in Patients with MDD in Adults (Study 1)

Schizophrenia
The efficacy of REXULTI in the treatment of adults with schizophrenia was demonstrated in two 6-week randomized, double-blind, placebo-controlled, fixed-dose clinical studies in patients who met DSM-IV-TR criteria for schizophrenia.
In both studies, Study 3 (NCT01396421) and Study 4 (NCT01393613), patients were randomized to REXULTI 2 or 4 mg once per day or placebo. Patients in the REXULTI groups initiated treatment at 1 mg once daily on Days 1 to 4. The REXULTI dosage was increased to 2 mg on Days 5 to 7. The dosage was then either maintained at 2 mg once daily or increased to 4 mg once daily, depending on treatment assignment, for the 5 remaining weeks.
The primary efficacy endpoint of both studies was the change from baseline to Week 6 in the Positive and Negative Syndrome Scale (PANSS) total score. The PANSS is a 30-item scale that measures positive symptoms of schizophrenia (7 items), negative symptoms of schizophrenia (7 items), and general psychopathology (16 items), each rated on a scale of 1 (absent) to 7 (extreme); the total PANSS scores range from 30 (best) to 210 (worst).
In Study 3, REXULTI at both 2 mg once daily and 4 mg once daily was superior to placebo on the PANSS total score. In Study 4, REXULTI 4 mg once daily was superior to placebo on the PANSS total score (Table 14). Figure 5 shows the time course of response based on the primary efficacy measure (change from baseline in PANSS total score) in Study 3.
Examination of population subgroups based on age, sex, and race did not suggest differential responsiveness.
| Study | Treatment Group | N | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference Difference (drug minus placebo) in least-squares mean change from baseline (95% CI) |
|---|---|---|---|---|---|
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: unadjusted confidence interval | |||||
| 3 | REXULTI (2 mg/day) Dosages statistically significantly superior to placebo | 180 | 95.9 (13.8) | -20.7 (1.5) | -8.7 (-13.1, -4.4) |
| REXULTI (4 mg/day) | 178 | 94.7 (12.1) | -19.7 (1.5) | -7.6 (-12.0, -3.1) | |
| Placebo | 178 | 95.7 (11.5) | -12.0 (1.6) | -- | |
| 4 | REXULTI (2 mg/day) | 179 | 96.3 (12.9) | -16.6 (1.5) | -3.1 (-7.2, 1.1) |
| REXULTI (4 mg/day) | 181 | 95.0 (12.4) | -20.0 (1.5) | -6.5 (-10.6, -2.4) | |
| Placebo | 180 | 94.6 (12.8) | -13.5 (1.5) | -- | |
Figure 5 Change from Baseline in PANSS Total Score by Study Visit (Week) in Adult Patients with Schizophrenia (Study 3)

The safety and efficacy of REXULTI as maintenance treatment in adults with schizophrenia aged 18 to 65 years were demonstrated in the maintenance phase of a randomized withdrawal study (Study 5, NCT01668797). Patients were stabilized for at least 12 weeks on 1 to 4 mg/day of REXULTI (N=202). They were then randomized in the double-blind treatment phase to either continue REXULTI at their achieved stable dose (N=97), or to switch to placebo (N=105).
The primary endpoint in Study 5 was time from randomization to impending relapse during the double-blind phase, defined as: 1) Clinical Global Improvement score of ≥5 (minimally worse) and an increase to a score >4 on PANSS conceptual disorganization, hallucinatory behavior, suspiciousness, or unusual thought content items, with either a ≥2 increase on a specific item or ≥4 point increase on the combined four PANSS items, 2) hospitalization due to worsening of psychotic symptoms, 3) current suicidal behavior, or 4) violent/aggressive behavior.
A pre-specified interim analysis demonstrated a statistically significantly longer time to relapse in patients randomized to the REXULTI group compared to placebo-treated patients. The study was subsequently terminated early because maintenance of efficacy had been demonstrated. The Kaplan-Meier curves of the cumulative proportion of patients with relapse during the double-blind treatment phase for REXULTI and placebo groups are shown in Figure 6. The key secondary endpoint, the proportion of patients who met the criteria for impending relapse, was statistically significantly lower in REXULTI-treated patients compared with placebo group.
Figure 6 Kaplan-Meier Estimation of Percent Impending Relapse in Study 5
| Note: A total of 202 patients were randomized. Among them, one patient in the placebo group did not take investigational medicinal product and one patient in the REXULTI group did not have post-randomization efficacy evaluations. These two patients were excluded from the efficacy analysis. |
 |
Pediatric Patients Ages 13 to 17 years
The efficacy of REXULTI in the treatment of schizophrenia in pediatric patients 13 to 17 years of age was demonstrated in a 6-week, randomized and placebo-controlled, clinical study.
In Study 6 (NCT03198078), patients were randomized to REXULTI 2 mg to 4 mg once per day, active comparator, or placebo. Patients in the REXULTI group initiated treatment at 0.5 mg once daily on Days 1 to 4. The REXULTI dosage was increased to 1 mg daily on Days 5 to 7 and then increased to 2 mg on Days 8 to 14. The dosage was then either maintained at 2 mg or increased to 3 mg once daily from Days 15 to 21 based on patient's tolerability or clinical response. After the titration period, patients were either kept at a maintenance dose, or increased or decreased by 1 mg, for a maximum of REXULTI 4 mg daily.
The primary efficacy endpoint was the change from baseline to Week 6 in the PANSS total score.
In Study 6, REXULTI group showed a statistically significant improvement compared to placebo on the mean change from baseline in the PANSS total score (Table 15).
Figure 7 shows the time course of response based on the primary efficacy measure (change from baseline in PANSS total score) in Study 6.
| Study | Treatment Group | N Efficacy sample includes treated subjects who have baseline and at least 1 post-baseline efficacy evaluation for the PANSS Total Score | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference Difference (drug minus placebo) in least-squares mean change from baseline (95% CI) |
|---|---|---|---|---|---|
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval | |||||
| 6 | REXULTI (2 - 4 mg/day) Dosages statistically significantly superior to placebo | 110 | 101.1 (14.9) | -22.8 (1.5) | -5.3 (-9.6, -1.1) |
| Placebo | 103 | 102.2 (16.3) | -17.4 (1.6) | -- | |
Figure 7 Change from Baseline in PANSS Total Score by Study Visit (Week) in Pediatric Patients 13 to 17 years of age with Schizophrenia (Study 6)

Agitation Associated with Dementia Due to Alzheimer's Disease
The efficacy of REXULTI in the treatment of agitation associated with dementia due to Alzheimer's disease was demonstrated in two 12-week, randomized, double-blind, placebo-controlled, fixed-dose studies (Study 7, NCT01862640 and Study 8, NCT03548584). In these studies, patients were required to:
- Have a diagnosis of probable Alzheimer's disease according to NINCDS-ADRDA criteria,
- Have a Mini-Mental State Examination (MMSE) score of ≥5 and ≤22 and have a total score of ≥4 by the agitation/aggression item of the NPI/NPI-NH, and
- Exhibit sufficient agitation behaviors at time of entry to warrant use of pharmacotherapy, after excluding other factors.
Patients in:
- Study 7 were randomized to an oral dosage of either REXULTI 1 mg once a day, REXULTI 2 mg once a day, or placebo. Patients in both REXULTI groups started on 0.25 mg once daily for approximately three days, then received 0.5 mg once daily for approximately 12 days. Subsequently, patients in the 1 mg group received 1 mg once daily for the remainder of the 12-week study, and patients in the 2 mg group received 1 mg once daily for approximately two weeks and then received 2 mg for the remainder of the study.
- Study 8 were randomized to an oral dose of either REXULTI 2 mg or 3 mg once a day (combined treatment arm) or placebo. Patients in both REXULTI groups started on 0.5 mg once daily for 7 days, then received 1 mg once daily for 7 days and then 2 mg once daily for 14 days. Subsequently, patients in the 2 mg group received 2 mg once daily for the remainder of the 12-week study, and patients in the 3 mg group received 3 mg once daily for the remainder of the study.
Study 7 included 433 patients with a mean age of 74 years old, and a range of 51 and 90 years old; 45% were male; 96%, 3%, and 1%, were White, Black or African American, and Asian, respectively; and 16% and 83% were Latino/Hispanic and not Latino/Hispanic, respectively. Study 8 included 345 patients with a mean age of 74 years old, and a range of 56 and 90 years old; 44% were male; 95%, 4%, and 1% were White, Black or African American, and Asian, respectively; and 31% and 69% were Latino/Hispanic and not Latino/Hispanic, respectively.
The primary efficacy endpoint in these two studies was the change from baseline in the Cohen-Mansfield Agitation Inventory total (CMAI) score at Week 12. The CMAI is a clinician rated questionnaire consisting of 29 items, which assess the frequency of manifestations of agitated behaviors in elderly patients, based on caregiver input. Three specific factors can be derived from the CMAI scale: 1) Aggressive Behavior (e.g., screaming, throwing things, cursing/verbal aggression, kicking, pushing scratching, hurting self or others); 2) Physically Non-Aggressive Behavior (e.g., repetitive mannerisms, general restlessness, pacing); and 3) Verbally Agitated Behavior (e.g., complaining, repetitive questions, constant requests for attention). Each CMAI behavior was rated on a scale of 1 (never) to 7 (very frequent agitated behaviors); the total CMAI scores range from 29 (best) to 203 (worst). A negative change indicates improvement.
In Trial 7, patients in the REXULTI 2 mg group showed improved total CMAI scores compared to patients in the placebo group at Week 12. In Trial 7, patients in the REXULTI 2 mg/3 mg group showed improved total CMAI scores compared to patients in the placebo group at Week 12.
As shown in Table 16 and Figure 8, the mean change from baseline in the total CMAI score after 12 weeks in the 2 mg/or 3 mg REXULTI group was statistically significantly superior to the placebo group. The 1 mg REXULTI group did not demonstrate significantly greater mean changes at baseline from the placebo group in the total CMAI score in this patient population. The 1 mg once day REXULTI dosage is not approved and is not recommended for the treatment of agitation associated with dementia due to Alzheimer's disease [see Dosage and Administration (2.4) ].
| Study | Treatment Group | N | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference Difference (drug minus placebo) in least-squares mean change from baseline (95% CI) |
|---|---|---|---|---|---|
| SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: unadjusted confidence interval | |||||
| 7 | REXULTI 1 mg/day | 134 | 70.5 (16.0) | -17.6 (1.3) | 0.2 (-3.4, 3.9) |
| REXULTI 2 mg/day Dosages statistically significantly superior to placebo. | 138 | 71.0 (16.6) | -21.6 (1.3) | -3.8 (-7.4, -0.2) | |
| Placebo | 131 | 72.2 (17.9) | -17.8 (1.3) | — | |
| 8 | REXULTI 2 mg/day or 3 mg/day | 225 | 80.6 (16.6) | -22.6 (1.1) | -5.3 (-8.8, -1.9) |
| Placebo | 116 | 79.2 (17.5) | -17.3 (1.4) | — | |
Figure 8 Change from Baseline in Total CMAI Score by Study Week in Patients with Agitation Associated with Dementia Due to Alzheimer's Disease (Study 8)
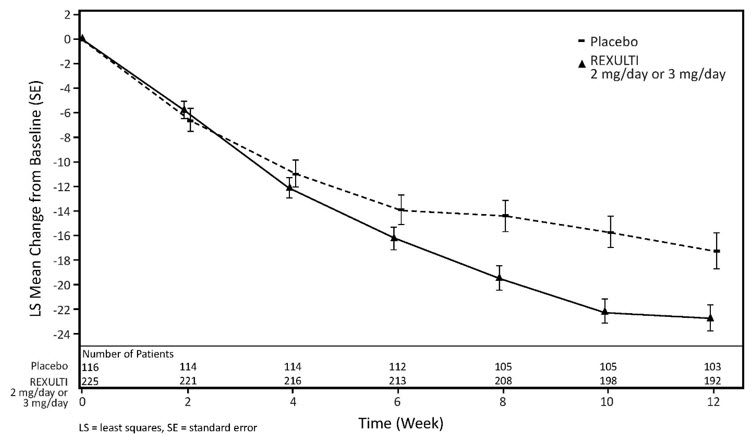
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
REXULTI (brexpiprazole) tablets have markings on one side and are available in the following strengths and package configurations (see below):
- 0.25 mg tablets are light brown, round, shallow convex, bevel-edged body with "BRX" and "0.25" imprinted on one side
NDC 59148-035-13 Bottles of 30
- 0.5 mg tablets: are light orange, round, shallow convex, bevel-edged body with "BRX" and "0.5" imprinted on one side
NDC 59148-036-13 Bottles of 30
- 1 mg tablets are light yellow, round, shallow convex, bevel-edged body with "BRX" and "1" imprinted on one side
NDC 59148-037-13 Bottles of 30
- 2 mg tablets are light green, round, shallow convex, bevel-edged body with "BRX" and "2" imprinted on one side
NDC 59148-038-13 Bottles of 30
- 3 mg tablets are light purple, round, shallow convex, bevel-edged body with "BRX" and "3" imprinted on one side
NDC 59148-039-13 Bottles of 30
- 4 mg tablets are white, round, shallow convex, bevel-edged body with "BRX" and "4" imprinted on one side
NDC 59148-040-13 Bottles of 30
Storage and Handling
Store REXULTI tablets at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].