Get your patient on Felbamate - Felbamate suspension (Felbamate)
Felbamate - Felbamate suspension prescribing information
BOXED WARNING
1.000000000000000e+00 APLASTIC ANEMIA
THE USE OF FELBAMATE IS ASSOCIATED WITH A MARKED INCREASE IN THE INCIDENCE OF APLASTIC ANEMIA. ACCORDINGLY, FELBAMATE SHOULD ONLY BE USED IN PATIENTS WHOSE EPILEPSY IS SO SEVERE THAT THE RISK OF APLASTIC ANEMIA IS DEEMED ACCEPTABLE IN LIGHT OF THE BENEFITS CONFERRED BY ITS USE (SEE INDICATIONS ). ORDINARILY, A PATIENT SHOULD NOT BE PLACED ON AND/OR CONTINUED ON FELBAMATE WITHOUT CONSIDERATION OF APPROPRIATE EXPERT HEMATOLOGIC CONSULTATION.
AMONG FELBAMATE TREATED PATIENTS, APLASTIC ANEMIA (PANCYTOPENIA IN THE PRESENCE OF A BONE MARROW LARGELY DEPLETED OF HEMATOPOIETIC PRECURSORS) OCCURS AT AN INCIDENCE THAT MAY BE MORE THAN A 100 FOLD GREATER THAN THAT SEEN IN THE UNTREATED POPULATION (I.E., 2 TO 5 PER MILLION PERSONS PER YEAR). THE RISK OF DEATH IN PATIENTS WITH APLASTIC ANEMIA GENERALLY VARIES AS A FUNCTION OF ITS SEVERITY AND ETIOLOGY; CURRENT ESTIMATES OF THE OVERALL CASE FATALITY RATE ARE IN THE RANGE OF 20 TO 30%, BUT RATES AS HIGH AS 70% HAVE BEEN REPORTED IN THE PAST.
THERE ARE TOO FEW FELBAMATE ASSOCIATED CASES, AND TOO LITTLE KNOWN ABOUT THEM TO PROVIDE A RELIABLE ESTIMATE OF THE SYNDROME'S INCIDENCE OR ITS CASE FATALITY RATE OR TO IDENTIFY THE FACTORS, IF ANY, THAT MIGHT CONCEIVABLY BE USED TO PREDICT WHO IS AT GREATER OR LESSER RISK.
IN MANAGING PATIENTS ON FELBAMATE, IT SHOULD BE BORNE IN MIND THAT THE CLINICAL MANIFESTATION OF APLASTIC ANEMIA MAY NOT BE SEEN UNTIL AFTER A PATIENT HAS BEEN ON FELBAMATE FOR SEVERAL MONTHS (E.G., ONSET OF APLASTIC ANEMIA AMONG FELBAMATE EXPOSED PATIENTS FOR WHOM DATA ARE AVAILABLE HAS RANGED FROM 5 TO 30 WEEKS). HOWEVER, THE INJURY TO BONE MARROW STEM CELLS THAT IS HELD TO BE ULTIMATELY RESPONSIBLE FOR THE ANEMIA MAY OCCUR WEEKS TO MONTHS EARLIER. ACCORDINGLY, PATIENTS WHO ARE DISCONTINUED FROM FELBAMATE REMAIN AT RISK FOR DEVELOPING ANEMIA FOR A VARIABLE, AND UNKNOWN, PERIOD AFTERWARDS.
IT IS NOT KNOWN WHETHER OR NOT THE RISK OF DEVELOPING APLASTIC ANEMIA CHANGES WITH DURATION OF EXPOSURE. CONSEQUENTLY, IT IS NOT SAFE TO ASSUME THAT A PATIENT WHO HAS BEEN ON FELBAMATE WITHOUT SIGNS OF HEMATOLOGIC ABNORMALITY FOR LONG PERIODS OF TIME IS WITHOUT RISK.
IT IS NOT KNOWN WHETHER OR NOT THE DOSE OF FELBAMATE AFFECTS THE INCIDENCE OF APLASTIC ANEMIA.
IT IS NOT KNOWN WHETHER OR NOT CONCOMITANT USE OF ANTIEPILEPTIC DRUGS AND/OR OTHER DRUGS AFFECTS THE INCIDENCE OF APLASTIC ANEMIA.
APLASTIC ANEMIA TYPICALLY DEVELOPS WITHOUT PREMONITORY CLINICAL OR LABORATORY SIGNS, THE FULL BLOWN SYNDROME PRESENTING WITH SIGNS OF INFECTION, BLEEDING, OR ANEMIA. ACCORDINGLY, ROUTINE BLOOD TESTING CANNOT BE RELIABLY USED TO REDUCE THE INCIDENCE OF APLASTIC ANEMIA, BUT, IT WILL, IN SOME CASES, ALLOW THE DETECTION OF THE HEMATOLOGIC CHANGES BEFORE THE SYNDROME DECLARES ITSELF CLINICALLY. FELBAMATE SHOULD BE DISCONTINUED IF ANY EVIDENCE OF BONE MARROW DEPRESSION OCCURS.
2.000000000000000e+00 HEPATIC FAILURE
EVALUATION OF POSTMARKETING EXPERIENCE SUGGESTS THAT ACUTE LIVER FAILURE IS ASSOCIATED WITH THE USE OF FELBAMATE. THE REPORTED RATE IN THE U.S. HAS BEEN ABOUT 6 CASES OF LIVER FAILURE LEADING TO DEATH OR TRANSPLANT PER 75,000 PATIENT YEARS OF USE. THIS RATE IS AN UNDERESTIMATE BECAUSE OF UNDER REPORTING, AND THE TRUE RATE COULD BE CONSIDERABLY GREATER THAN THIS. FOR EXAMPLE, IF THE REPORTING RATE IS 10%, THE TRUE RATE WOULD BE ONE CASE PER 1,250 PATIENT YEARS OF USE.
OF THE CASES REPORTED, ABOUT 67% RESULTED IN DEATH OR LIVER TRANSPLANTATION, USUALLY WITHIN 5 WEEKS OF THE ONSET OF SIGNS AND SYMPTOMS OF LIVER FAILURE. THE EARLIEST ONSET OF SEVERE HEPATIC DYSFUNCTION FOLLOWED SUBSEQUENTLY BY LIVER FAILURE WAS 3 WEEKS AFTER INITIATION OF FELBAMATE. ALTHOUGH SOME REPORTS DESCRIBED DARK URINE AND NONSPECIFIC PRODROMAL SYMPTOMS (E.G., ANOREXIA, MALAISE, AND GASTROINTESTINAL SYMPTOMS), IN OTHER REPORTS IT WAS NOT CLEAR IF ANY PRODROMAL SYMPTOMS PRECEDED THE ONSET OF JAUNDICE.
IT IS NOT KNOWN WHETHER OR NOT THE RISK OF DEVELOPING HEPATIC FAILURE CHANGES WITH DURATION OF EXPOSURE.
IT IS NOT KNOWN WHETHER OR NOT THE DOSAGE OF FELBAMATE AFFECTS THE INCIDENCE OF HEPATIC FAILURE.
IT IS NOT KNOWN WHETHER CONCOMITANT USE OF OTHER ANTIEPILEPTIC DRUGS AND/OR OTHER DRUGS AFFECT THE INCIDENCE OF HEPATIC FAILURE.
FELBAMATE SHOULD NOT BE PRESCRIBED FOR ANYONE WITH A HISTORY OF HEPATIC DYSFUNCTION.
TREATMENT WITH FELBAMATE SHOULD BE INITIATED ONLY IN INDIVIDUALS WITHOUT ACTIVE LIVER DISEASE AND WITH NORMAL BASELINE SERUM TRANSAMINASES. IT HAS NOT BEEN PROVED THAT PERIODIC SERUM TRANSAMINASE TESTING WILL PREVENT SERIOUS INJURY BUT IT IS GENERALLY BELIEVED THAT EARLY DETECTION OF DRUGINDUCED HEPATIC INJURY ALONG WITH IMMEDIATE WITHDRAWAL OF THE SUSPECT DRUG ENHANCES THE LIKELIHOOD FOR RECOVERY. THERE IS NO INFORMATION AVAILABLE THAT DOCUMENTS HOW RAPIDLY PATIENTS CAN PROGRESS FROM NORMAL LIVER FUNCTION TO LIVER FAILURE, BUT OTHER DRUGS KNOWN TO BE HEPATOTOXINS CAN CAUSE LIVER FAILURE RAPIDLY (E.G., FROM NORMAL ENZYMES TO LIVER FAILURE IN 2-4 WEEKS). ACCORDINGLY, MONITORING OF SERUM TRANSAMINASE LEVELS (AST AND ALT) IS RECOMMENDED AT BASELINE AND PERIODICALLY THEREAFTER. WHILE THE MORE FREQUENT THE MONITORING THE GREATER THE CHANCES OF EARLY DETECTION, THE PRECISE SCHEDULE FOR MONITORING IS A MATTER OF CLINICAL JUDGEMENT.
FELBAMATE SHOULD BE DISCONTINUED IF EITHER SERUM AST OR SERUM ALT LEVELS BECOME INCREASED ≥ 2 TIMES THE UPPER LIMIT OF NORMAL, OR IF CLINICAL SIGNS AND SYMPTOMS SUGGEST LIVER FAILURE (SEE PRECAUTIONS). PATIENTS WHO DEVELOP EVIDENCE OF HEPATOCELLULAR INJURY WHILE ON FELBAMATE AND ARE WITHDRAWN FROM THE DRUG FOR ANY REASON SHOULD BE PRESUMED TO BE AT INCREASED RISK FOR LIVER INJURY IF FELBAMATE IS REINTRODUCED. ACCORDINGLY, SUCH PATIENTS SHOULD NOT BE CONSIDERED FOR RE-TREATMENT.
INDICATIONS & USAGE
Felbamate oral suspension is not indicated as a first line antiepileptic treatment (see Warnings ). Felbamate oral suspension is recommended for use only in those patients who respond inadequately to alternative treatments and whose epilepsy is so severe that a substantial risk of aplastic anemia and/or liver failure is deemed acceptable in light of the benefits conferred by its use.
If these criteria are met and the patient has been fully advised of the risk, and has provided written acknowledgement, felbamate oral suspension can be considered for either monotherapy or adjunctive therapy in the treatment of partial seizures, with and without generalization, in adults with epilepsy and as adjunctive therapy in the treatment of partial and generalized seizures associated with Lennox-Gastaut syndrome in children.
DOSAGE & ADMINISTRATION
Felbamate oral suspension has been studied as monotherapy and adjunctive therapy in adults and as adjunctive therapy in children with seizures associated with Lennox-Gastaut syndrome. As felbamate oral suspension is added to or substituted for existing AEDs, it is strongly recommended to reduce the dosage of those AEDs in the range of 20-33% to minimize side effects (see Drug Interactions subsection).
Dosage Adjustment in the Renally Impaired: Felbamate should be used with caution in patients with renal dysfunction. In the renally impaired, starting and maintenance doses should be reduced by one-half (see CLINICAL PHARMACOLOGY/Pharmacokinetics and PRECAUTIONS ). Adjunctive therapy with medications which affect felbamate plasma concentrations, especially AEDs, may warrant further reductions in felbamate daily doses in patients with renal dysfunction.
Adults (14 years of age and over)
The majority of patients received 3600 mg/day in clinical trials evaluating its use as both monotherapy and adjunctive therapy.
Monotherapy: (Initial therapy) felbamate oral suspension has not been systematically evaluated as initial monotherapy. Initiate felbamate oral suspension at 1200 mg/day in divided doses three or four times daily. The prescriber is advised to titrate previously untreated patients under close clinical supervision, increasing the dosage in 600-mg increments every 2 weeks to 2400 mg/day based on clinical response and thereafter to 3600 mg/day if clinically indicated.
Conversion to Monotherapy: Initiate felbamate oral suspension at 1200 mg/day in divided doses three or four times daily. Reduce the dosage of concomitant AEDs by one-third at initiation of felbamate oral suspension therapy. At week 2, increase the felbamate oral suspension dosage to 2400 mg/day while reducing the dosage of other AEDs up to an additional one-third of their original dosage. At week 3, increase the felbamate dosage up to 3600 mg/day and continue to reduce the dosage of other AEDs as clinically indicated.
Adjunctive Therapy: Felbamate should be added at 1200 mg/day in divided doses three or four times daily while reducing present AEDs by 20% in order to control plasma concentrations of concurrent phenytoin, valproic acid, phenobarbital, and carbamazepine and its metabolites. Further reductions of the concomitant AEDs dosage may be necessary to minimize side effects due to drug interactions. Increase the dosage of felbamate by 1200 mg/day increments at weekly intervals to 3600 mg/day. Most side effects seen during felbamate adjunctive therapy resolve as the dosage of concomitant AEDs is decreased.
| Table 6 Dosage Table (adults) | |||
| Dosage reduction of concomitant AEDs | WEEK 1 REDUCE original dose by 20–33%• | WEEK 2 REDUCE original dose by up to an additional 1/3• | WEEK 3 REDUCE as clinically indicated |
| Felbamate Dosage | 1200 mg/dayInitial dose | 2400 mg/day Therapeutic dosage range | 3600 mg/day Therapeutic dosage range |
| •See Adjunctive and Conversion to Monotherapy sections. | |||
While the above felbamate conversion guidelines may result in a felbamate 3600 mg/day dose within 3 weeks, in some patients titration to a 3600 mg/day felbamate dose has been achieved in as little as 3 days with appropriate adjustment of other AEDs.
Children with Lennox-Gastaut Syndrome (Ages 2-14 years)
Adjunctive Therapy: Felbamate should be added at 15 mg/kg/day in divided doses three or four times daily while reducing present AEDs by 20% in order to control plasma levels of concurrent phenytoin, valproic acid, phenobarbital, and carbamazepine and its metabolites. Further reductions of the concomitant AEDs dosage may be necessary to minimize side effects due to drug interactions. Increase the dosage of felbamate by 15 mg/kg/day increments at weekly intervals to 45 mg/kg/day. Most side effects seen during felbamate adjunctive therapy resolve as the dosage of concomitant AEDs is decreased.
CONTRAINDICATIONS
Felbamate oral suspension is contraindicated in patients with known hypersensitivity to felbamate, its ingredients, or known sensitivity to other carbamates. It should not be used in patients with a history of any blood dyscrasia or hepatic dysfunction.
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Novitium Pharma LLC at 1-855-204-1431 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
The most common adverse reactions seen in association with felbamate in adults during monotherapy are anorexia, vomiting, insomnia, nausea, and headache. The most common adverse reactions seen in association with felbamate in adults during adjunctive therapy are anorexia, vomiting, insomnia, nausea, dizziness, somnolence, and headache.
The most common adverse reactions seen in association with felbamate in children during adjunctive therapy are anorexia, vomiting, insomnia, headache, and somnolence.
The dropout rate because of adverse experiences or intercurrent illnesses among adult felbamate patients was 12 percent (120/977). The dropout rate because of adverse experiences or intercurrent illnesses among pediatric felbamate patients was six percent (22/357). In adults, the body systems associated with causing these withdrawals in order of frequency were: digestive (4.3%), psychological (2.2%), whole body (1.7%), neurological (1.5%), and dermatological (1.5%). In children, the body systems associated with causing these withdrawals in order of frequency were: digestive (1.7%), neurological (1.4%), dermatological (1.4%), psychological (1.1%), and whole body (1.0%). In adults, specific events with an incidence of 1% or greater associated with causing these withdrawals, in order of frequency were: anorexia (1.6%), nausea (1.4%), rash (1.2%), and weight decrease (1.1%). In children, specific events with an incidence of 1% or greater associated with causing these withdrawals, in order of frequency was rash (1.1%).
Incidence in Clinical Trials:
The prescriber should be aware that the figures cited in the following table cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different investigators, treatments, and uses including the use of felbamate as adjunctive therapy where the incidence of adverse events may be higher due to drug interactions. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contribution of drug and nondrug factors to the side effect incidence rate in the population studied.
Adults
Incidence in Controlled Clinical Trials-Monotherapy Studies in Adults:
The table that follows enumerates adverse events that occurred at an incidence of 2% or more among 58 adult patients who received felbamate monotherapy at dosages of 3600 mg/day in double-blind controlled trials. Table 3 presents reported adverse events that were classified using standard WHO-based dictionary terminology.
| Table 3 Adults Treatment-Emergent Adverse Event Incidence in Controlled Monotherapy Trials | ||
| Felbamate• (N=58) | Low Dose Valproate•• (N=50) | |
| Body System Event | % | % |
| Body as a Whole | ||
| Fatigue | 6.9 | 4.0 |
| Weight Decrease | 3.4 | 0 |
| Face Edema | 3.4 | 0 |
| Central Nervous System | ||
| Insomnia | 8.6 | 4.0 |
| Headache | 6.9 | 18.0 |
| Anxiety | 5.2 | 2.0 |
| Dermatological | ||
| Acne | 3.4 | 0 |
| Rash | 3.4 | 0 |
| Digestive | ||
| Dyspepsia | 8.6 | 2.0 |
| Vomiting | 8.6 | 2.0 |
| Constipation | 6.9 | 2.0 |
| Diarrhea | 5.2 | 0 |
| SGPT Increased | 5.2 | 2.0 |
| Metabolic/Nutritional | ||
| Hypophosphatemia | 3.4 | 0 |
| Respiratory | ||
| Upper Respiratory Tract Infection | 8.6 | 4.0 |
| Rhinitis | 6.9 | 0 |
| Special Senses | ||
| Diplopia | 3.4 | 4.0 |
| Otitis Media | 3.4 | 0 |
| Urogenital | ||
| Intramenstrual Bleeding | 3.4 | 0 |
| Urinary Tract Infection | 3.4 | 2.0 |
| •3600 mg/day;•• 15 mg/kg/day | ||
Incidence in Controlled Add-On Clinical Studies in Adults:
Table 4 enumerates adverse events that occurred at an incidence of 2% or more among 114 adult patients who received felbamate adjunctive therapy in add-on controlled trials at dosages up to 3600 mg/day. Reported adverse events were classified using standard WHO-based dictionary terminology.
Many adverse experiences that occurred during adjunctive therapy may be a result of drug interactions. Adverse experiences during adjunctive therapy typically resolved with conversion to monotherapy, or with adjustment of the dosage of other antiepileptic drugs.
| Table 4 Adults Treatment-Emergent Adverse Event Incidence in Controlled Add-On Trials | ||
| Felbamate | Placebo | |
| (N=114) | (N=43) | |
| Body System/Event | % | % |
| Body as a Whole | ||
| Fatigue | 16.8 | 7.0 |
| Fever | 2.6 | 4.7 |
| Chest Pain | 2.6 | 0 |
| Central Nervous System | ||
| Headache | 36.8 | 9.3 |
| Somnolence | 19.3 | 7.0 |
| Dizziness | 18.4 | 14.0 |
| Insomnia | 17.5 | 7.0 |
| Nervousness | 7.0 | 2.3 |
| Tremor | 6.1 | 2.3 |
| Anxiety | 5.3 | 4.7 |
| Gait Abnormal | 5.3 | 0 |
| Depression | 5.3 | 0 |
| Paraesthesia | 3.5 | 2.3 |
| Ataxia | 3.5 | 0 |
| Mouth Dry | 2.6 | 0 |
| Stupor | 2.6 | 0 |
| Dermatological | ||
| Rash | 3.5 | 4.7 |
| Digestive | ||
| Nausea | 34.2 | 2.3 |
| Anorexia | 19.3 | 2.3 |
| Vomiting | 16.7 | 4.7 |
| Dyspepsia | 12.3 | 7.0 |
| Constipation | 11.4 | 2.3 |
| Diarrhea | 5.3 | 2.3 |
| Abdominal Pain | 5.3 | 0 |
| SGPT Increased | 3.5 | 0 |
| Musculoskeletal | ||
| Myalgia | 2.6 | 0 |
| Respiratory | ||
| Upper Respiratory Tract Infection | 5.3 | 7.0 |
| Sinusitis | 3.5 | 0 |
| Pharyngitis | 2.6 | 0 |
| Special Senses | ||
| Diplopia | 6.1 | 0 |
| Taste Perversion | 6.1 | 0 |
| Vision Abnormal | 5.3 | 2.3 |
Children
Incidence in a Controlled Add-On Trial in Children with Lennox-Gastaut Syndrome:
Table 5 enumerates adverse events that occurred more than once among 31 pediatric patients who received felbamate up to 45 mg/kg/day or a maximum of 3600 mg/day. Reported adverse events were classified using standard WHO-based dictionary terminology.
| Table 5 Children Treatment-Emergent Adverse Event Incidence in Controlled Add-On Lennox-Gastaut Trials | ||
| Felbamate | Placebo | |
| (N=31) | (N=27) | |
| Body System/Event | % | % |
| Body as a Whole Fever Fatigue Weight Decrease Pain | 22.6 9.7 6.5 6.5 | 11.1 3.7 0 0 |
| Central Nervous System | ||
| Somnolence | 48.4 | 11.1 |
| Insomnia | 16.1 | 14.8 |
| Nervousness | 16.1 | 18.5 |
| Gait Abnormal | 9.7 | 0 |
| Headache | 6.5 | 18.5 |
| Thinking Abnormal | 6.5 | 3.7 |
| Ataxia | 6.5 | 3.7 |
| Urinary Incontinence | 6.5 | 7.4 |
| Emotional Lability | 6.5 | 0 |
| Miosis | 6.5 | 0 |
| Dermatological | ||
| Rash | 9.7 | 7.4 |
| Digestive | ||
| Anorexia | 54.8 | 14.8 |
| Vomiting | 38.7 | 14.8 |
| Constipation | 12.9 | 0 |
| Hiccup | 9.7 | 3.7 |
| Nausea | 6.5 | 0 |
| Dyspepsia | 6.5 | 3.7 |
| Hematologic | ||
| Purpura | 12.9 | 7.4 |
| Leukopenia | 6.5 | 0 |
| Respiratory | ||
| Upper Respiratory Tract Infection | 45.2 | 25.9 |
| Pharyngitis | 9.7 | 3.7 |
| Coughing | 6.5 | 0 |
| Special Senses | ||
| Otitis Media | 9.7 | 0 |
Other Events Observed in Association with the Administration of Felbamate:
In the paragraphs that follow, the adverse clinical events, other than those in the preceding tables, that occurred in a total of 977 adults and 357 children exposed to felbamate and that are reasonably associated with its use are presented. They are listed in order of decreasing frequency. Because the reports cite events observed in open-label and uncontrolled studies, the role of felbamate in their causation cannot be reliably determined.
Events are classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring on one or more occasions in at least 1/100 patients; infrequent adverse events are those occurring in 1/100-1/1000 patients; and rare events are those occurring in fewer than 1/1000 patients.
Event frequencies are calculated as the number of patients reporting an event divided by the total number of patients (N=1334) exposed to felbamate.
Body as a Whole : Frequent: Weight increase, asthenia, malaise, influenza-like symptoms; Rare: anaphylactoid reaction, chest pain substernal.
Cardiovascular : Frequent: Palpitation, tachycardia; Rare: supraventricular tachycardia.
Central Nervous System : Frequent: Agitation, psychological disturbance, aggressive reaction;
Infrequent: hallucination, euphoria, suicide attempt, migraine.
Digestive : Frequent: SGOT increased; Infrequent: esophagitis, appetite increased; Rare: GGT elevated.
Hematologic : Infrequent: Lymphadenopathy, leukopenia, leukocytosis, thrombocytopenia, granulocytopenia; Rare: antinuclear factor test positive, qualitative platelet disorder, agranulocytosis.
Metabolic/Nutritional : Infrequent: Hypokalemia, hyponatremia, LDH increased, alkaline phosphatase increased, hypophosphatemia; Rare: creatinine phosphokinase increased.
Musculoskeletal : Infrequent: Dystonia.
Dermatological : Frequent: Pruritus; Infrequent: urticaria, bullous eruption; Rare: buccal mucous membrane swelling, Stevens-Johnson Syndrome.
Special Senses : Rare: Photosensitivity allergic reaction.
Postmarketing Adverse Event Reports:
Voluntary reports of adverse events in patients taking felbamate (usually in conjunction with other drugs) have been received since market introduction and may have no causal relationship with the drug(s). These include the following by body system:
Body as a Whole : neoplasm, sepsis, L.E. syndrome, SIDS, sudden death, edema, hypothermia, rigors, hyperpyrexia.
Cardiovascular : atrial fibrillation, atrial arrhythmia, cardiac arrest, torsade de pointes, cardiac failure, hypotension, hypertension, flushing, thrombophlebitis, ischemic necrosis, gangrene, peripheral ischemia, bradycardia, Henoch-Schönlein purpura (vasculitis).
Central & Peripheral Nervous System : delusion, paralysis, mononeuritis, cerebrovascular disorder, cerebral edema, coma, manic reaction, encephalopathy, paranoid reaction, nystagmus, choreoathetosis, extrapyramidal disorder, confusion, psychosis, status epilepticus, dyskinesia, dysarthria, respiratory depression, apathy, concentration impaired.
Dermatological : abnormal body odor, sweating, lichen planus, livedo reticularis, alopecia, toxic
epidermal necrolysis.
Digestive : (Refer to WARNINGS ) hepatitis, hepatic failure, G.I. hemorrhage, hyperammonemia, pancreatitis, hematemesis, gastritis, rectal hemorrhage, flatulence, gingival bleeding, acquired megacolon, ileus, intestinal obstruction, enteritis, ulcerative stomatitis, glossitis, dysphagia, jaundice, gastric ulcer, gastric dilatation, gastroesophageal reflux.
Fetal Disorders : fetal death, microcephaly, genital malformation, anencephaly, encephalocele.
Hematologic : (Refer to WARNINGS ) increased and decreased prothrombin time, anemia, hypochromic anemia, aplastic anemia, pancytopenia, hemolytic uremic syndrome, increased mean corpuscular volume (mcv) with and without anemia, coagulation disorder, embolism-limb, disseminated intravascular coagulation, eosinophilia, hemolytic anemia, leukemia, including myelogenous leukemia, and lymphoma, including T-cell and B-cell lymphoproliferative disorders.
Metabolic/Nutritional : hypernatremia, hypoglycemia, SIADH, hypomagnesemia, dehydration,
hyperglycemia, hypocalcemia.
Musculoskeletal : arthralgia, muscle weakness, involuntary muscle contraction, rhabdomyolysis.
Respiratory : dyspnea, pneumonia, pneumonitis, hypoxia, epistaxis, pleural effusion, respiratory insufficiency, pulmonary hemorrhage, asthma.
Special Senses : hemianopsia, decreased hearing, conjunctivitis.
Urogenital : menstrual disorder, acute renal failure, hepatorenal syndrome, hematuria, urinary retention, nephrosis, vaginal hemorrhage, abnormal renal function, dysuria, placental disorder.
DESCRIPTION
Felbamate, USP is an antiepileptic available as a 600 mg/5 mL suspension for oral administration. Its chemical name is 2-phenyl-1,3-propanediol dicarbamate.
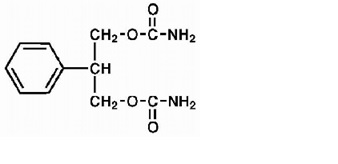
Felbamate, USP is a white to off-white crystalline powder with a characteristic odor. It is very slightly soluble in water, slightly soluble in ethanol, sparingly soluble in methanol, and freely soluble in dimethyl sulfoxide. The molecular weight is 238.24; felbamate, USP’s molecular formula is C 11 H 14 N 2 O 4 ; its structural formula is:
The inactive ingredients for Felbamate Oral Suspension, USP 600 mg/5 mL are noncrystallizing sorbitol solution, microcrystalline cellulose and carboxymethylcellulose sodium, glycerin, methylparaben, propylparaben, polysorbate 80, simethicone emulsion, saccharin sodium monohydrate, bubblegum flavor (contains arabic gum, and natural and artificial flavor), FD&C Red No. 40, FD&C Yellow No. 6, and purified water.
CLINICAL PHARMACOLOGY
Mechanism of Action:
The mechanism by which felbamate exerts its anticonvulsant activity is unknown, but in animal test systems designed to detect anticonvulsant activity, felbamate has properties in common with other marketed anticonvulsants. Felbamate is effective in mice and rats in the maximal electroshock test, the subcutaneous pentylenetetrazol seizure test, and the subcutaneous picrotoxin seizure test. Felbamate also exhibits anticonvulsant activity against seizures induced by intracerebroventricular administration of glutamate in rats and N-methyl-D,L-aspartic acid in mice. Protection against maximal electroshockinduced seizures suggests that felbamate may reduce seizure spread, an effect possibly predictive of efficacy in generalized tonic-clonic or partial seizures. Protection against pentylenetetrazol-induced seizures suggests that felbamate may increase seizure threshold, an effect considered to be predictive of potential efficacy in absence seizures.
Receptor-binding studies in vitro indicate that felbamate has weak inhibitory effects on GABA-receptor binding, benzodiazepine receptor binding, and is devoid of activity at the MK-801 receptor binding site of the NMDA receptor-ionophore complex. However, felbamate does interact as an antagonist at the strychnine-insensitive glycine recognition site of the NMDA receptor-ionophore complex. Felbamate is not effective in protecting chick embryo retina tissue against the neurotoxic effects of the excitatory amino acid agonists NMDA, kainate, or quisqualate in vitro.
The monocarbamate, p-hydroxy, and 2-hydroxy metabolites were inactive in the maximal electroshockinduced seizure test in mice. The monocarbamate and p-hydroxy metabolites had only weak (0.2 to 0.6) activity compared with felbamate in the subcutaneous pentylenetetrazol seizure test. These metabolites did not contribute significantly to the anticonvulsant action of felbamate.
Pharmacokinetics:
The numbers in the pharmacokinetic section are mean ± standard deviation.
Felbamate is well-absorbed after oral administration. Over 90% of the radioactivity after a dose of 1000 mg 14 C felbamate was found in the urine. Absolute bioavailability (oral vs. parenteral) has not been measured. The tablet and suspension were each shown to be bioequivalent to the capsule used in clinical trials, and pharmacokinetic parameters of the tablet and suspension are similar. There was no effect of food on absorption of the tablet; the effect of food on absorption of the suspension has not been evaluated.
Following oral administration, felbamate is the predominant plasma species (about 90% of plasma radioactivity). About 40-50% of absorbed dose appears unchanged in urine, and an additional 40% is present as unidentified metabolites and conjugates. About 15% is present as parahydroxyfelbamate, 2hydroxyfelbamate, and felbamate monocarbamate, none of which have significant anticonvulsant activity.
Binding of felbamate to human plasma protein was independent of felbamate concentrations between 10 and 310 micrograms/mL. Binding ranged from 22% to 25%, mostly to albumin, and was dependent on the albumin concentration.
Felbamate is excreted with a terminal half-life of 20-23 hours, which is unaltered after multiple doses. Clearance after a single 1200 mg dose is 26±3 mL/hr/kg, and after multiple daily doses of 3600 mg is 30±8 mL/hr/kg. The apparent volume of distribution was 756±82 mL/kg after a 1200 mg dose. Felbamate C max and AUC are proportionate to dose after single and multiple doses over a range of 100-800 mg single doses and 1200-3600 mg daily doses. C min (trough) blood levels are also dose proportional. Multiple daily doses of 1200, 2400, and 3600 mg gave C min values of 30±5, 55±8, and 83±21 micrograms/mL (N=10 patients). Linear and dose proportional pharmacokinetics were also observed at doses above 3600 mg/day up to the maximum dose studied of 6000 mg/day. Felbamate gave dose proportional steady-state peak plasma concentrations in children age 4-12 over a range of 15, 30, and 45 mg/kg/day with peak concentrations of 17, 32, and 49 micrograms/mL.
The effects of race and gender on felbamate pharmacokinetics have not been systematically evaluated, but plasma concentrations in males (N=5) and females (N=4) given felbamate have been similar. The effects of felbamate kinetics on hepatic functional impairment have not been evaluated.
Renal Impairment:
Felbamate’s single dose monotherapy pharmacokinetic parameters were evaluated in 12 otherwise healthy individuals with renal impairment. There was a 40-50% reduction in total body clearance and 9-15 hours prolongation of half-life in renally impaired subjects compared to that in subjects with normal renal function. Reduced felbamate clearance and a longer half-life were associated with diminishing renal function.
Pharmacodynamics:
Typical Physiologic Responses:
- Cardiovascular:
In adults, there is no effect of felbamate on blood pressure. Small but statistically significant mean increases in heart rate were seen during adjunctive therapy and monotherapy; however, these mean increases of up to 5 bpm were not clinically significant. In children, no clinically relevant changes in blood pressure or heart rate were seen during adjunctive therapy or monotherapy with felbamate.
2.000000000000000e+00 Other Physiologic Effects:
The only other change in vital signs was a mean decrease of approximately one (1) respiration per minute in respiratory rate during adjunctive therapy in children. In adults, statistically significant mean reductions in body weight were observed during felbamate monotherapy and adjunctive therapy. In children, there were mean decreases in body weight during adjunctive therapy and monotherapy; however, these mean changes were not statistically significant. These mean reductions in adults and children were approximately 5% of the mean weights at baseline.
CLINICAL STUDIES
The results of controlled clinical trials established the efficacy of felbamate as monotherapy and adjunctive therapy in adults with partial-onset seizures with or without secondary generalization and in partial and generalized seizures associated with Lennox-Gastaut syndrome in children.
Felbamate Monotherapy Trials in Adults
Felbamate (3600 mg/day given four times-a-day) and low-dose valproate (15 mg/kg/day) were compared as monotherapy during a 112-day treatment period in a multicenter and a single-center double-blind efficacy trial. Both trials were conducted according to an identical study design. During a 56-day baseline period, all patients had at least four partial-onset seizures per 28 days and were receiving one antiepileptic drug at a therapeutic level, the most common being carbamazepine. In the multicenter trial, baseline seizure frequencies were 12.4 per 28 days in the felbamate group and 21.3 per 28 days in the low-dose valproate group. In the single-center trial, baseline seizure frequencies were 18.1 per 28 days in the felbamate group and 15.9 per 28 days in the low-dose valproate group. Patients were converted to monotherapy with felbamate or low-dose valproic acid during the first 28 days of the 112-day treatment period. Study endpoints were completion of 112 study days or fulfilling an escape criterion. Criteria for escape relative to baseline were: (1) twofold increase in monthly seizure frequency, (2) twofold increase in highest 2-day seizure frequency, (3) single generalized tonic-clonic seizure (GTC) if none occurred during baseline, or (4) significant prolongation of GTCs. The primary efficacy variable was the number of patients in each treatment group who met escape criteria.
In the multicenter trial, the percentage of patients who met escape criteria was 40% (18/45) in the felbamate group and 78% (39/50) in the low-dose valproate group. In the single-center trial, the percentage of patients who met escape criteria was 14% (3/21) in the felbamate group and 90% (19/21) in the low-dose valproate group. In both trials, the difference in the percentage of patients meeting escape criteria was statistically significant (P<.001) in favor of felbamate. These two studies by design were intended to demonstrate the effectiveness of felbamate monotherapy. The studies were not designed or intended to demonstrate comparative efficacy of the two drugs. For example, valproate was not used at the maximally effective dose.
Felbamate Adjunctive Therapy Trials in Adults
A double-blind, placebo-controlled crossover trial consisted of two 10-week outpatient treatment periods. Patients with refractory partial-onset seizures who were receiving phenytoin and carbamazepine at therapeutic levels were administered felbamate as add-on therapy at a starting dosage of 1400 mg/day in three divided doses, which was increased to 2600 mg/day in three divided doses. Among the 56 patients who completed the study, the baseline seizure frequency was 20 per month. Patients treated with felbamate had fewer seizures than patients treated with placebo for each treatment sequence. There was a 23% (P=.018) difference in percentage seizure frequency reduction in favor of felbamate.
Felbamate 3600 mg/day given four times-a-day and placebo were compared in a 28-day double-blind add-on trial in patients who had their standard antiepileptic drugs reduced while undergoing evaluations for surgery of intractable epilepsy. All patients had confirmed partial-onset seizures with or without generalization, seizure frequency during surgical evaluation not exceeding an average of four partial seizures per day or more than one generalized seizure per day, and a minimum average of one partial or generalized tonicclonic seizure per day for the last 3 days of the surgical evaluation. The primary efficacy variable was time to fourth seizure after randomization to treatment with felbamate or placebo. Thirteen (46%) of 28 patients in the felbamate group versus 29 (88%) of 33 patients in the placebo group experienced a fourth seizure. The median times to fourth seizure were greater than 28 days in the felbamate group and 5 days in the placebo group. The difference between felbamate and placebo in time to fourth seizure was statistically significant (P=.002) in favor of felbamate.
Felbamate Adjunctive Therapy Trial in Children with Lennox-Gastaut Syndrome
In a 70-day double-blind, placebo-controlled add-on trial in the Lennox-Gastaut syndrome, felbamate 45 mg/kg/day given four times-a-day was superior to placebo in controlling the multiple seizure types associated with this condition. Patients had at least 90 atonic and/or atypical absence seizures per month while receiving therapeutic dosages of one or two other antiepileptic drugs. Patients had a past history of using an average of eight antiepileptic drugs. The most commonly used antiepileptic drug during the baseline period was valproic acid. The frequency of all types of seizures during the baseline period was 1617 per month in the felbamate group and 716 per month in the placebo group. Statistically significant differences in the effect on seizure frequency favored felbamate over placebo for total seizures (26% reduction vs. 5% increase, P<.001), atonic seizures (44% reduction vs. 7% reduction, P=.002), and generalized tonic-clonic seizures (40% reduction vs. 12% increase, P=.017). Parent/guardian global evaluations based on impressions of quality of life with respect to alertness, verbal responsiveness, general well-being, and seizure control significantly (P<.001) favored felbamate over placebo.
When efficacy was analyzed by gender in four well-controlled trials of felbamate as adjunctive and monotherapy for partial-onset seizures and Lennox-Gastaut syndrome, a similar response was seen in 122 males and 142 females.
HOW SUPPLIED
Felbamate Oral Suspension, USP 600 mg/5 mL, is a pink colored homogeneous suspension free from visible agglomeration with bubblegum odor available in 8 oz bottles (NDC 70954-051-10) and 16 oz bottles (NDC 70954-051-20).
Shake suspension well before using. Store oral suspension at 20° to 25°C (68° to 77°F); excursions permitted between 15° to 30°C (59° to 86°F). [See USP Controlled Room Temperature].
Dispense in a tight container.
To report SUSPECTED ADVERSE REACTIONS, contact Novitium Pharma LLC at 1-855-204-1431 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
Manufactured by:
Novitium Pharma LLC
70 Lake Drive, East Windsor
New Jersey 08520
Issued: 05/2022
LB4039-03