Get your patient on Methylergonovine Maleate - Methylergonovine Maleate tablet (Methylergonovine Maleate)
Methylergonovine Maleate - Methylergonovine Maleate tablet prescribing information
INDICATIONS AND USAGE
Following delivery of the placenta, for routine management of uterine atony, hemorrhage and subinvolution of the uterus. For control of uterine hemorrhage in the second stage of labor following delivery of the anterior shoulder.
DOSAGE AND ADMINISTRATION
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.
Orally
One tablet, 0.2 mg, 3 or 4 times daily in the puerperium for a maximum of 1 week.
CONTRAINDICATIONS
Hypertension; toxemia; pregnancy; and hypersensitivity.
ADVERSE REACTIONS
The most common adverse reaction is hypertension associated in several cases with seizure and/or headache. Hypotension has also been reported. Abdominal pain (caused by uterine contractions), nausea and vomiting have occurred occasionally. Rarely observed reactions have included: acute myocardial infarction, transient chest pains, vasoconstriction, vasospasm, coronary arterial spasm, bradycardia, tachycardia, dyspnea, hematuria, thrombophlebitis, water intoxication, hallucinations, leg cramps, dizziness, tinnitus, nasal congestion, diarrhea, diaphoresis, palpitation, rash, and foul taste.
There have been rare isolated reports of anaphylaxis, without a proven causal relationship to the drug product.
Postmarketing Experience
The following adverse drug reactions have been derived from post-marketing experience with methylergonovine maleate via spontaneous case reports. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency which is therefore categorized as not known.
Nervous system disorders
Cerebrovascular accident, paraesthesia
Cardiac disorders
Ventricular fibrillation, ventricular tachycardia, angina pectoris, atrioventricular block
To report SUSPECTED ADVERSE REACTIONS, contact Teva at 1-888-838-2872 or FDA at 1-800-FDA-1088 or http://www.fda.gov/medwatch.
Drug Interactions
CYP 3A4 Inhibitors (e.g., Macrolide Antibiotics and Protease Inhibitors)
There have been rare reports of serious adverse events in connection with the coadministration of certain ergot alkaloid drugs (e.g., dihydroergotamine and ergotamine) and potent CYP 3A4 inhibitors, resulting in vasospasm leading to cerebral ischemia and/or ischemia of the extremities. Although there have been no reports of such interactions with methylergonovine alone, potent CYP 3A4 inhibitors should not be coadministered with methylergonovine. Examples of some of the more potent CYP 3A4 inhibitors include macrolide antibiotics (e.g., erythromycin, troleandomycin, clarithromycin), HIV protease or reverse transcriptase inhibitors (e.g., ritonavir, indinavir, nelfinavir, delavirdine) or azole antifungals (e.g., ketoconazole, itraconazole, voriconazole). Less potent CYP 3A4 inhibitors should be administered with caution. Less potent inhibitors include saquinavir, nefazodone, fluconazole, grapefruit juice, fluoxetine, fluvoxamine, zileuton, and clotrimazole. These lists are not exhaustive, and the prescriber should consider the effects on CYP 3A4 of other agents being considered for concomitant use with methylergonovine.
CYP3A4 inducers
Drugs (e.g. nevirapine, rifampicin) that are strong inducers of CYP3A4 are likely to decrease the pharmacological action of methylergonovine maleate.
Beta-blockers
Caution should be exercised when methylergonovine maleate is used concurrently with beta-blockers. Concomitant administration with beta-blockers may enhance the vasoconstrictive action of ergot alkaloids.
Anesthetics
Anesthetics like halothan and methoxyfluran may reduce the oxytocic potency of methylergonovine maleate.
Glyceryl trinitrate and other antianginal drugs
Methylergonovine maleate produces vasoconstriction and can be expected to reduce the effect of glyceryl trinitrate and other antianginal drugs.
No pharmacokinetic interactions involving other cytochrome P450 isoenzymes are known.
Caution should be exercised when methylergonovine maleate is used concurrently with other vasoconstrictors, ergot alkaloids, or prostaglandins.
DESCRIPTION
Methylergonovine maleate is a semi-synthetic ergot alkaloid used for the prevention and control of postpartum hemorrhage.
Methylergonovine maleate is available in tablets for oral ingestion containing 0.2 mg methylergonovine maleate.
Active Ingredient: methylergonovine maleate USP, 0.2 mg. Inactive Ingredients: acacia, gelatin, lactose monohydrate, methylparaben, microcrystalline cellulose, povidone, propylparaben, corn starch, stearic acid, and tartaric acid.
Chemically, methylergonovine maleate is designated as ergoline-8-carboxamide, 9,10-didehydro- N -[1- (hydroxymethyl)propyl]-6-methyl-, [8β( S )]-, ( Z )-2-butenedioate (1:1) (salt).
Its structural formula is
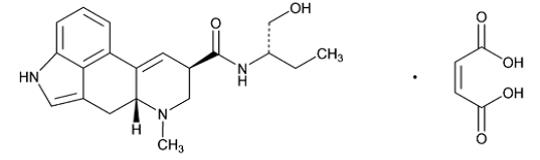
C 20 H 25 N 3 O 2 ·C 4 H 4 O 4 M.W. 455.50
FDA approved dissolution test specifications differ from USP.
CLINICAL PHARMACOLOGY
Methylergonovine maleate acts directly on the smooth muscle of the uterus and increases the tone, rate, and amplitude of rhythmic contractions. Thus, it induces a rapid and sustained tetanic uterotonic effect which shortens the third stage of labor and reduces blood loss. The onset of action after I.V. administration is immediate; after I.M. administration, 2 to 5 minutes, and after oral administration, 5 to 10 minutes.
Pharmacokinetic studies following an I.V. injection have shown that methylergonovine is rapidly distributed from plasma to peripheral tissues within 2 to 3 minutes or less. The bioavailability after oral administration was reported to be about 60% with no accumulation after repeated doses. During delivery, with intramuscular injection, bioavailability increased to 78%. Ergot alkaloids are mostly eliminated by hepatic metabolism and excretion, and the decrease in bioavailability following oral administration is probably a result of first-pass metabolism in the liver.
Bioavailability studies conducted in fasting healthy female volunteers have shown that oral absorption of a 0.2 mg methylergonovine tablet was fairly rapid with a mean peak plasma concentration of 3243 ± 1308 pg/mL observed at 1.12 ± 0.82 hours. For a 0.2 mg intramuscular injection, a mean peak plasma concentration of 5918 ± 1952 pg/mL was observed at 0.41 ± 0.21 hours. The extent of absorption of the tablet, based upon methylergonovine plasma concentrations, was found to be equivalent to that of the I.M. solution given orally, and the extent of oral absorption of the I.M. solution was proportional to the dose following administration of 0.1, 0.2, and 0.4 mg. When given intramuscularly, the extent of absorption of methylergonovine solution was about 25% greater than the tablet. The volume of distribution (Vdss/F) of methylergonovine was calculated to be 56.1 ± 17.0 liters, and the plasma clearance (CLp/F) was calculated to be 14.4 ± 4.5 liters per hour. The plasma level decline was biphasic with a mean elimination half-life of 3.39 hours (range 1.5 to 12.7 hours). A delayed gastrointestinal absorption (T max about 3 hours) of methylergonovine tablet might be observed in postpartum women during continuous treatment with this oxytocic agent.
HOW SUPPLIED
Methylergonovine maleate tablets USP, 0.2 mg are available as white to off white, round shaped tablets, debossed with “TV” on one side and “2J” on the other side containing 0.2 mg methylergonovine maleate, USP packaged in bottles of 12 tablets (NDC 0093- 3655 -22) and 28 tablets (NDC 0093- 3655 -28).
Store and Dispense
Store at 20˚ to 25˚C (68˚ to 77˚F) [see USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
Keep this and all medications out of the reach of children.
Manufactured In Israel By:
Teva Pharmaceutical Ind. Ltd.
Kfar Saba, 4410202, Israel
Manufactured For:
Teva Pharmaceuticals
Parsippany, NJ 07054
Rev. A 12/2024