Get your patient on Nulibry - Fosdenopterin Hydrobromide injection, Powder, Lyophilized, For Solution (Fosdenopterin Hydrobromide)
Nulibry - Fosdenopterin Hydrobromide injection, Powder, Lyophilized, For Solution prescribing information
INDICATIONS AND USAGE
NULIBRY is indicated to reduce the risk of mortality in patients with molybdenum cofactor deficiency (MoCD) Type A.
DOSAGE AND ADMINISTRATION
- Start NULIBRY if known or presumed MoCD Type A. Promptly discontinue if MoCD Type A is not confirmed by genetic testing. (2.1 )
- NULIBRY is intended for administration by a healthcare provider. If deemed appropriate by a healthcare provider, NULIBRY may be administered at home by the patient's caregiver. (2.2 )
- See the table below for the recommended dosage in patients less than one year of age. Administer NULIBRY by intravenous infusion once daily. For dose volumes less than 2 mL, consider administering by slow intravenous push. (2.3 )
| Titration Duration | Preterm Neonates (Gestational Age Less than 37 Weeks) | Term Neonates (Gestational Age 37 weeks and Above) |
| Initial Dosage | 0.4 mg/kg once daily | 0.55 mg/kg once daily |
| Dosage at Month 1 and Month 2 | 0.7 mg/kg once daily | 0.75 mg/kg once daily |
| Dosage at Month 3 to Month 12 | 0.9 mg/kg once daily | 0.9 mg/kg once daily |
- Recommended Dosage in Patients One Year of Age or Older: 0.9 mg/kg given as an intravenous infusion once daily. (2.3 )
Patient Selection
Start NULIBRY if the patient has a diagnosis or presumptive diagnosis of MoCD Type A.
In patients with a presumptive diagnosis of MoCD Type A, confirm the diagnosis of MoCD Type A immediately after initiation of NULIBRY treatment. In such patients, discontinue NULIBRY if the MoCD Type A diagnosis is not confirmed by genetic testing.
Important Recommendation Prior to NULIBRY Treatment
NULIBRY is intended for administration by a healthcare provider. If deemed appropriate by a healthcare provider, NULIBRY may be administered at home by the patient's caregiver. If NULIBRY can be administered by a caregiver/patient, advise them to read the detailed instructions on the preparation, administration, storage, and disposal of NULIBRY for caregivers [see Instructions for Use ].
Recommended Dosage and Administration
Recommended Dosage and Administration in Patients Less Than One Year of Age (by gestational age)
The recommended dosage regimen of NULIBRY in patients less than one year of age (by gestational age), is based on actual body weight as shown in Table 1 . Administer NULIBRY by intravenous infusion once daily. For dose volumes less than 2 mL, consider administering by slow intravenous push using syringe administration [ see Dosage and Administration (2.4 ) ] .
| Titration Duration | Preterm Neonates (Gestational Age Less than 37 Weeks) | Term Neonates (Gestational Age 37 Weeks and Above) |
| Initial Dosage | 0.4 mg/kg once daily | 0.55 mg/kg once daily |
| Dosage at Month 1 and Month 2 | 0.7 mg/kg once daily | 0.75 mg/kg once daily |
| Dosage at Month 3 to Month 12 | 0.9 mg/kg once daily | 0.9 mg/kg once daily |
Recommended Dosage and Administration in Patients One Year of Age or Older
For patients one year of age or older, the recommended dosage of NULIBRY is 0.9 mg/kg (based on actual body weight) administered as an intravenous infusion once daily.
Missed Dose
If a NULIBRY dose is missed, administer the missed dose as soon as possible. Administer the next scheduled dose at least 6 hours after the administration of the missed dose.
Preparation and Administration Instructions
Preparation
NULIBRY must be reconstituted prior to use. Use aseptic technique during preparation and follow these instructions:
- Calculate the dose based on the patient's weight to determine the number of vials needed and total reconstituted dose volume. More than one vial may be needed to achieve the calculated dose.
- Remove the required number of vials from the freezer to allow them to reach room temperature (by hand warming for 3 to 5 minutes or exposing to ambient air for approximately 30 minutes).
- Reconstitute each NULIBRY vial with 5 mL of Sterile Water for Injection, USP.
- Gently swirl the vial continuously until the powder is completely dissolved. DO NOT shake. After reconstitution, the final concentration of NULIBRY reconstituted solution is 9.5 mg/5 mL (1.9 mg/mL).
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Reconstituted NULIBRY is a clear and colorless to pale yellow solution. Do not use if there are particles present or if the solution is discolored.
- Withdraw the calculated dose volume from the vial(s) using a syringe. Discard any unused solution remaining in the vial(s).
Administration
If reconstituted NULIBRY solution in the vial is refrigerated, allow it to come to room temperature (by hand warming for 3 to 5 minutes or exposing to ambient air for approximately 30 minutes) before administration.
- Administer NULIBRY as an intravenous infusion using an infusion pump at a rate of 1.5 mL per minute. Use non-DEHP tubing and a 0.2 micron filter to administer the total reconstituted dose volume.
- For dose volumes less than 2 mL, consider administering by slow intravenous push over 1 to 2 minutes using syringe administration.
- Complete administration of NULIBRY within 4 hours of reconstitution [ see Dosage and Administration (2.6 ) ] .
- Do not mix NULIBRY with other drugs. Do not administer as an infusion with other drugs.
Home Administration
If deemed appropriate by a healthcare provider, NULIBRY may be administered at home by the patient's caregiver. If NULIBRY can be administered by a caregiver/patient, advise them to read the detailed instructions on the preparation, administration, storage, and disposal of NULIBRY for caregivers [see Instructions for Use ].
Storage of Reconstituted Solution
- If the reconstituted NULIBRY solution in the vial is not used immediately, store at room temperature at 15°C to 25°C (59°F to 77°F) or refrigerate at 2°C to 8°C (36°F to 46°F) for up to 4 hours (inclusive of infusion time), or discard.
- Do not heat. Do not re-freeze NULIBRY after reconstitution. Do not shake.
DOSAGE FORMS AND STRENGTHS
For injection: 9.5 mg of fosdenopterin, as a white to pale yellow lyophilized powder or cake in a single-dose vial for reconstitution.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
There are no available data on NULIBRY use in pregnant females to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, no adverse developmental outcomes occurred at dose exposures 32-89 times the human exposure at the maximum recommended human dose (MRHD) of 0.9 mg/kg/day (see Data ).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryofetal development study, intravenous administration of fosdenopterin to pregnant rabbits once daily during the period of organogenesis (GD 7 to GD 20) at doses up to 200 mg/kg/day (approximately 89 times the human exposure at the MRHD, based on AUC) did not result in adverse developmental effects.
In a pre- and postnatal development study, subcutaneous administration of fosdenopterin to pregnant mice once daily throughout pregnancy and lactation (GD 6 to Lactation Day 20) at doses of 50, 100, or 200 mg/kg/day (up to 32 times the human exposure at the MRHD, based on AUC) did not result in adverse developmental effects.
Lactation
Risk Summary
There are no human or animal data available to assess the presence of NULIBRY or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production for the mother.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for NULIBRY and any potential adverse effects on the breastfed infant from NULIBRY or from the underlying maternal condition.
Pediatric Use
Safety and effectiveness of NULIBRY for the treatment of MoCD Type A have been established in pediatric patients starting from birth. Use of NULIBRY for this indication is supported by evidence from two open-label studies (Studies 1 and 2) and one observational study (Study 3), in which 13 pediatric patients aged birth to 6 years of age were treated with NULIBRY or rcPMP. Pediatric use information is discussed throughout the labeling.
Animal studies have identified that NULIBRY has phototoxic potential. Advise NULIBRY-treated patients or their caregivers to avoid patient exposure to direct sunlight and artificial UV light exposure (i.e., UVA or UVB phototherapy) and adopt precautionary measures [ see Warnings and Precautions (5.1 ) and Nonclinical Toxicology (13.2 ) ].
Geriatric Use
MoCD Type A is largely a disease of pediatric patients. Clinical studies of NULIBRY did not include patients 65 years of age and older.
Adult Use
The safety and effectiveness of NULIBRY for the treatment of adults with MoCD Type A have been established. Use of NULIBRY in adults for this indication is based on an adequate and well- controlled clinical investigation in pediatric patients [ see Clinical Studies (14 ) ].
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Potential for Photosensitivity : Advise patients/caregivers to avoid patient exposure to sunlight, and to have the patient wear sunscreen, protective clothing, and sunglasses when exposed to the sun. If photosensitivity occurs, advise caregivers/patients to seek medical attention immediately and consider a dermatological evaluation. (5.1 , 13.2 )
Potential for Photosensitivity
Animal studies have identified that NULIBRY has phototoxic potential [ see Nonclinical Toxicology (13.2 ) ].
Advise NULIBRY-treated patients or their caregivers to avoid or minimize patient exposure to direct sunlight and artificial UV light exposure (i.e., UVA or UVB phototherapy) and adopt precautionary measures (e.g., have the patient wear protective clothing and hats, use broad spectrum sunscreen with high sun protection factor (SPF) in patients 6 months of age and older, and wear sunglasses when exposed to the sun). If photosensitivity occurs, advise caregivers/patients to seek medical attention immediately and consider a dermatological evaluation.
ADVERSE REACTIONS
The most common adverse reactions (>25%) were catheter-related complications, pyrexia, viral infection, pneumonia, otitis media, vomiting, cough/sneezing, viral upper respiratory infection, gastroenteritis, bacteremia, and diarrhea. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Sentynl Therapeutcis, Inc. at 888-507-5206 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Overview of Safety Evaluation
The safety of NULIBRY was assessed in 37 pediatric patients and healthy adults who received at least one intravenous infusion of NULIBRY or an E. coli derived non-salt, anhydrous form of cPMP (recombinant cPMP or rcPMP, which has the same active moiety and therefore the same biologic activity as NULIBRY). Of these 37 patients/healthy adults, 13 were pediatric patients with MoCD Type A in Studies 1, 2, and 3 [ see Clinical Studies (14 )] , 6 were pediatric patients with presumptive MoCD Type A but who were later confirmed to not have MoCD Type A, and 18 were healthy adults (without MoCD Type A) in a Phase 1 study.
Adverse Reactions
Assessment of adverse reactions for NULIBRY is based on data from two open-label, single-arm studies, Study 1 (n=8) and Study 2 (n=1), in patients with a confirmed diagnosis of MoCD Type A (8 of the 9 patients were previously treated with rcPMP). In these studies, patients received a daily intravenous infusion of NULIBRY. The median exposure to NULIBRY was 4.3 years and ranged from 8 days to 5.6 years [ see Clinical Studies (14 )]. In these studies, 44% of patients were males and 56% were females, 67% were White and 33% were Asian. The mean age was 14 days and ranged from 1 day to 69 days at time of first infusion.
Table 2 presents the most common adverse reactions that occurred in NULIBRY-treated patients in Studies 1 and 2.
| Adverse Reactions | NULIBRY-Treated Patients (N=9) n (%) |
|---|---|
Abbreviations: MoCD = molybdenum cofactor deficiency | |
1 Catheter-related complications included complication associated with device, catheter site abscess, catheter site discharge, catheter site extravasation, catheter site pain, catheter site infection, catheter site inflammation, device dislocation, device leakage, device occlusion, and vascular device infection. | |
| Catheter-related complications 1 | 8 (89%) |
| Pyrexia | 7 (78%) |
| Viral infection | 5 (56%) |
| Pneumonia | 4 (44%) |
| Otitis Media | 4 (44%) |
| Vomiting | 4 (44%) |
| Cough/Sneezing | 4 (44%) |
| Upper viral respiratory infection | 3 (33%) |
| Gastroenteritis | 3 (33%) |
| Diarrhea | 3 (33%) |
| Bacteremia | 3 (33%) |
| Abdominal pain | 2 (22%) |
| Influenza | 2 (22%) |
| Lower respiratory tract infection | 2 (22%) |
| Viral tonsillitis | 2 (22%) |
| Oropharyngeal pain | 2 (22%) |
| Rash maculo-papular | 2 (22%) |
| Anemia | 2 (22%) |
| Eye swelling | 2 (22%) |
| Seizure | 2 (22%) |
| Agitation | 2 (22%) |
Safety data are also available from 10 patients with MoCD Type A who received rcPMP in Study 3 (an observational study) [ see Clinical Studies (14 )]. The median time on rcPMP treatment was 1.5 years and ranged from 6 days to 4.4 years. In Study 3, the patient population was evenly distributed between males and females with a mean age of 18 days (range 1, 69) at time of first infusion, 70% were White, and 30% were Asian.
In Study 3, one patient died of necrotizing enterocolitis. Adverse reactions for the rcPMP-treated patients were similar to the NULIBRY-treated patients, except for the following additional adverse reactions that were reported in more than one patient: sepsis, oral candidiasis, varicella, fungal skin infection, and eczema.
DESCRIPTION
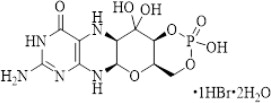
NULIBRY (fosdenopterin) for injection is cyclic pyranopterin monophosphate (cPMP). Fosdenopterin is present as a dihydrate of the hydrobromide salt with the chemical name (4a R ,5a R ,11a R ,12a S )-8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-1,3,2-dioxaphosphorino[4',5':5,6]pyrano[3,2- g ]pteridin-10(4 H )-one 2-oxide. Fosdenopterin hydrobromide as a dihydrate is a crystalline solid. The molecular formula is C 10 H 14 N 5 O 8 P • HBr • 2H 2 O and the molecular weight is 480.16. The chemical structure is:
NULIBRY is supplied as a sterile, preservative-free, white to pale yellow lyophilized powder or cake in a single-dose, clear glass vial for reconstitution for intravenous infusion. Each vial contains 9.5 mg fosdenopterin (equivalent to 12.5 mg fosdenopterin hydrobromide as a dihydrate). Each vial also contains the following inactive ingredients: 10 mg ascorbic acid USP, 187.5 mg mannitol USP, and 62.5 mg sucrose NF. Sodium hydroxide NF and hydrochloric acid NF are used to adjust pH to 5.0-7.0.
CLINICAL PHARMACOLOGY
Mechanism of Action
Patients with MoCD Type A have mutations in the MOCS1 gene leading to deficient MOCS1A/B dependent synthesis of the intermediate substrate, cPMP. Substrate replacement therapy with NULIBRY provides an exogenous source of cPMP, which is converted to molybdopterin. Molybdopterin is then converted to molybdenum cofactor, which is needed for the activation of molybdenum-dependent enzymes, including sulfite oxidase (SOX), an enzyme that reduces levels of neurotoxic sulfites.
Pharmacodynamics
In MoCD Type A, the lack of effective SOX leads to elevated levels of the neurotoxic sulfite, S-sulfocysteine (SSC). Treatment with NULIBRY resulted in a reduction in the level of urinary SSC normalized to creatinine and the reduction was sustained with long-term treatment with NULIBRY [ see Clinical Studies (14 ) ] .
Cardiac Electrophysiology
At the maximum approved recommended dose, NULIBRY did not prolong the QT interval to any clinically relevant extent.
Pharmacokinetics
The pharmacokinetics of fosdenopterin in healthy adult subjects following a single intravenous NULIBRY infusion are summarized in Table 3 . The area under the plasma concentration-time curve (AUC) and the maximum plasma concentration (C max ) of fosdenopterin increased in an approximately proportional manner with increasing doses.
1 0.075 mg/kg, 0.24 mg/kg, and 0.68 mg/kg doses are 0.08, 0.27, and 0.76 times the recommended maximum dose, respectively. | |||
| Parameter | 0.075 mg/kg 1 | 0.24 mg/kg 1 | 0.68 mg/kg 1 |
| C max (ng/mL) | 285 (57) | 873 (99) | 2800 (567) |
| AUC 0-inf (ng•h/mL) | 523 (75) | 1790 (213) | 5960 (1820) |
Distribution
The volume of distribution (V d ) of fosdenopterin was approximately 300 mL/kg. The plasma protein binding of fosdenopterin ranged from 6 to 12%.
Elimination
The mean total body clearance (CL) of fosdenopterin ranged from 167 to 195 mL/h/kg. The mean half-life of fosdenopterin ranged from 1.2 to 1.7 hours.
Metabolism
Fosdenopterin is predominantly metabolized through nonenzymatic degradation processes to Compound Z, an inactive oxidation product of endogenous cPMP.
Excretion
Renal clearance of fosdenopterin accounts for approximately 40% of total body clearance.
Specific Populations
The effect of renal and hepatic impairment on the pharmacokinetics of fosdenopterin is unknown.
Pediatric Patients
Pharmacokinetic properties of fosdenopterin in pediatric MoCD Type A patients are similar to healthy adult subjects.
Drug Interaction Studies
In Vitro Studies
Cytochrome P450 (CYP) Enzymes : Fosdenopterin does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5. Fosdenopterin does not induce CYP1A2, CYP2B6, or CYP3A4.
Transporter Systems : Fosdenopterin is a weak inhibitor of MATE2-K and OAT1, but does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OCT2, OAT3, and MATE1. Fosdenopterin is a weak substrate for MATE1, but is not a substrate of P-gp, BCRP, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, or MATE2-K.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies have not been conducted with fosdenopterin.
Mutagenesis
Fosdenopterin was not genotoxic in a standard battery of in vitro (bacterial reverse mutation and human lymphocyte chromosomal aberration) and in vivo (rodent bone marrow micronucleus) assays.
Impairment of Fertility
In a fertility and embryo-fetal development study, subcutaneous administration of fosdenopterin to mice once daily from 14 days prior to mating and continuing through GD 15 at doses up to 200 mg/kg/day (approximately 18 times the MRHD of 0.9 mg/kg/day, based on body surface area) did not adversely affect fertility or reproductive performance.
Animal Toxicology and/or Pharmacology
Fosdenopterin has demonstrated phototoxic potential in an animal study at doses equal to and greater than 4.5 times the maximum recommended human dose (based on human equivalent dose comparison). In this study, which was conducted in pigmented rats, intravenous (bolus) administration of fosdenopterin for three consecutive days followed by ultraviolet radiation (UVR) exposure resulted in dose-dependent cutaneous skin reactions (erythema, edema, flaking, and eschar) and ophthalmic and histopathologic changes indicative of phototoxicity [ see Warnings and Precautions (5.1 ) ].
CLINICAL STUDIES
The efficacy of NULIBRY for the treatment of patients with MoCD Type A was established based on data from three clinical studies (Studies 1, 2, and 3) that were compared to data from a natural history study.
Study 1
Study 1 (NCT02047461) was a prospective, open-label, single-arm, dose escalation study in patients with MoCD Type A who were receiving treatment with rcPMP prior to treatment with NULIBRY. Study 1 included 8 patients, 6 of whom previously participated in Study 3. The initial NULIBRY dosage was matched to the patient's rcPMP dosage upon entering the study. The NULIBRY dosage was then titrated over a period of 5 months to a maximum dosage of 0.9 mg/kg administered once daily as an intravenous infusion.
Study 2
Study 2 (NCT02629393) was a prospective, open-label, single-arm, dose escalation study in one patient with MoCD Type A who had not been previously treated with rcPMP. The initial dosage of NULIBRY in Study 2 was based on the gestational age of the patient (i.e., 36 weeks). The initial dosage was then incrementally escalated up to a maximum dosage of 0.98 mg/kg administered once daily as an intravenous infusion (1.1 times the maximum approved recommended dosage) [ see Dosage and Administration (2.3 ) ].
Study 3
Study 3 was a retrospective, observational study that included 10 patients with a confirmed diagnosis of MoCD Type A who received rcPMP. Six of these 10 patients were later enrolled in Study 1 to receive treatment with NULIBRY.
Efficacy Results
The efficacy of NULIBRY and rcPMP were assessed in a combined analysis of the 13 patients with genetically confirmed MoCD Type A from Study 1 (n=8), Study 2 (n=1), and Study 3 (n=4) who received substrate replacement therapy with NULIBRY or rcPMP.
Of the 13 treated patients included in the combined analysis, 54% were male, 77% were White and 23% were Asian; the median gestational age was 39 weeks (range 35 to 41 weeks). Of these 13 treated patients, the age at first dose was ≤ 14 days for 10 patients (with 5 patients initiating treatment at 1 day of age) and ≥ 32 days and < 69 days for the remaining 3 patients.
Overall Survival
Efficacy was assessed by comparing overall survival in pediatric patients treated with NULIBRY or rcPMP (n=13) with an untreated natural history cohort of pediatric patients with genetically confirmed MoCD Type A who were genotype-matched to the treated patients (n=18). Patients treated with NULIBRY or rcPMP had an improvement in overall survival compared to the untreated, genotype-matched, historical control group (Table 4 and Figure 1 ). Results were similar when comparing treated patients with all patients in the untreated natural history cohort with genetically confirmed MoCD Type A (n=37, includes the 18 genotype-matched untreated patients as well as 19 additional untreated patients who were not genotype-matched).
Abbreviations: CI=confidence interval; NE=not estimable; rcPMP=recombinant Escherichia coli -derived cPMP. | |||
a Quartile estimates from product-limit (Kaplan-Meier) method, with associated log-log confidence intervals. | |||
b Based on the area under the survival curves up to 1 year of follow-up. | |||
c Based on the area under the survival curves up to 3 years of follow-up. | |||
d Based on Cox proportional hazards model regressing survival status on an indicator variable denoting treatment status. The 95% CIs are based on the modified score test statistic under the Cox model. The hazard ratio represents the risk of death in the treated patients compared to the untreated historical control patients. | |||
| NULIBRY (or rcPMP) (n=13) | Untreated Genotype-Matched Historical Control (n=18) | Treatment Difference (95% CI) | |
| Number of Deaths (%) | 2 (15%) | 12 (67%) | |
| 50th Percentile (Median) Survival Time in Months (95% CI) a | NE (16, NE) months | 48 (10, 99) months | |
| Kaplan Meier Survival Probability (95% CI) | |||
| 1 year | 92% (57%, 99%) | 67% (40%, 83%) | |
| 3 years | 84% (49%, 96%) | 55% (30%, 74%) | |
| Mean Survival Time (Months) | |||
| At 1 year b (95% CI) | 11 (9, 13) months | 10 (8, 12) months | 1 (-1, 4) months |
| At 3 years c (95% CI) | 32 (26, 37) months | 24 (17, 31) months | 8 (-1, 16) months |
| Hazard Ratio for Risk of Death (95% CI) d | 0.18 (0.04, 0.72) | ||
Figure 1 Kaplan Meier Curve for Overall Survival in Patients with MoCD Type A Treated with NULIBRY or rcPMP Versus Genotype-Matched Untreated Patients in Historical Control
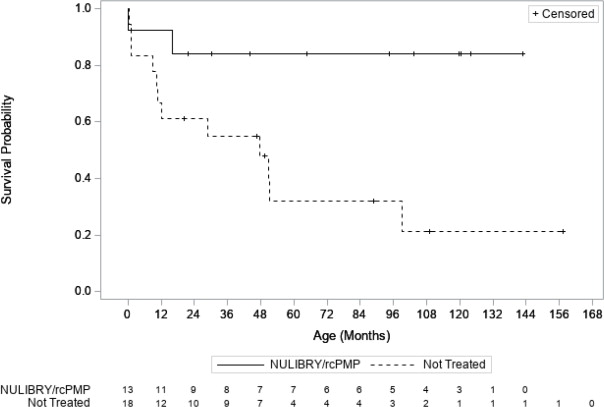
Abbreviations: rcPMP=recombinant Escherichia coli -derived cPMP
MoCD Biomarker Results
Treatment with NULIBRY resulted in a reduction in urine concentrations of SSC in patients with MoCD Type A and the reduction was sustained with long-term treatment over 48 months. The baseline level of urinary SSC normalized to creatinine was characterized in one patient (Study 2) with a value of 89.8 μmol/mmol. Following treatment with NULIBRY in Studies 1 and 2 (n=9), the mean ± SD levels of urinary SSC normalized to creatinine ranged from 11 (±8.5) to 7 (±2.4) μmol/mmol from Month 3 to Month 48.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
NULIBRY (fosdenopterin) for injection is a white to pale yellow lyophilized powder or cake in a single-dose clear glass vial for reconstitution. Each NULIBRY vial contains 9.5 mg of fosdenopterin.
Each carton of NULIBRY contains one vial (NDC 42358-295-01).
Components are not made with natural rubber latex.
Storage
Store NULIBRY frozen between -25°C and -10°C (-13°F and 14°F). Store vial in original carton to protect from light. For storage recommendations for the reconstituted solution [ see Dosage and Administration (2.6 ) ] .
Mechanism of Action
Patients with MoCD Type A have mutations in the MOCS1 gene leading to deficient MOCS1A/B dependent synthesis of the intermediate substrate, cPMP. Substrate replacement therapy with NULIBRY provides an exogenous source of cPMP, which is converted to molybdopterin. Molybdopterin is then converted to molybdenum cofactor, which is needed for the activation of molybdenum-dependent enzymes, including sulfite oxidase (SOX), an enzyme that reduces levels of neurotoxic sulfites.