Get your patient on Pirfenidone - Pirfenidone tablet, Coated (Pirfenidone)
Pirfenidone - Pirfenidone tablet, Coated prescribing information
Adverse Reactions (6.1 ) 2/2022
INDICATIONS AND USAGE
Pirfenidone is indicated for the treatment of idiopathic pulmonary fibrosis (IPF).
DOSAGE AND ADMINISTRATION
- Take with food.
- Recommended dosage: 801 mg three times daily (2403 mg/day). (2 )
- Upon initiation of treatment, titrate to the full dosage of 2403 mg/day over a 14-day period as follows:
| Treatment days | Dosage |
| Days 1 through 7 | 267 mg three times daily (801 mg/day) |
| Days 8 through 14 | 534 mg three times daily (1602 mg/day) |
| Days 15 onward | 801 mg three times daily (2403 mg/day) |
Testing Prior to Pirfenidone Tablets Administration
Conduct liver function tests prior to initiating treatment with pirfenidone [see Warnings and Precautions (5.1) ].
Recommended Dosage
The recommended daily maintenance dosage of pirfenidone is 801 mg three times daily for a total of 2403 mg/day. Doses should be taken with food at the same time each day.
Upon initiation of treatment, titrate to the full dosage of 2403 mg/day over a 14-day period as follows:
| Treatment days | Dosage |
| Days 1 through 7 | 267 mg three times daily (801 mg/day) |
| Days 8 through 14 | 534 mg three times daily (1602 mg/day) |
| Days 15 onward | 801 mg three times daily (2403 mg/day) |
Dosages above 2403 mg/day are not recommended for any patient. Patients should not take 2 doses at the same time to make up for a missed dose. Patients should not take more than 3 doses per day.
Dosage Modifications due to Adverse Reactions
Patients who miss 14 or more days of pirfenidone should re-initiate treatment by undergoing the initial 2-week titration regimen up to the full maintenance dosage [see Dosage and Administration (2.2) ]. For treatment interruption of less than 14 days, the dosage prior to the interruption can be resumed.
If patients experience significant adverse reactions (i.e., gastrointestinal, photosensitivity reaction or rash), consider temporary dosage reductions or interruptions of pirfenidone to allow for resolution of symptoms [see Warnings and Precautions (5.1 , 5.2 , 5.3) ].
Dosage Modification due to Elevated Liver Enzymes
Dosage modifications or interruptions may also be necessary when liver enzyme and bilirubin elevations are exhibited. For liver enzyme elevations, modify the dosage as follows:
If a patient exhibits >3 but ≤5 × the upper limit of normal (ULN) ALT and/or AST without symptoms or hyperbilirubinemia after starting pirfenidone therapy:
- Discontinue confounding medications, exclude other causes, and monitor the patient closely.
- Repeat liver chemistry tests as clinically indicated.
- The full daily dosage may be maintained, if clinically appropriate, or reduced or interrupted (e.g., until liver chemistry tests are within normal limits) with subsequent retitration to the full dosage as tolerated.
If a patient exhibits >3 but ≤5 × ULN ALT and/or AST accompanied by symptoms or hyperbilirubinemia:
- Permanently discontinue pirfenidone.
- Do not rechallenge patient with pirfenidone.
If a patient exhibits >5 × ULN ALT and/or AST:
- Permanently discontinue pirfenidone.
- Do not rechallenge patient with pirfenidone.
Dosage Modification due to Drug Interactions
Strong CYP1A2 Inhibitors (e.g., fluvoxamine, enoxacin)
Reduce pirfenidone to 267 mg three times a day (801 mg/day).
Moderate CYP1A2 Inhibitors (e.g., ciprofloxacin)
With use of ciprofloxacin at a dosage of 750 mg twice daily, reduce pirfenidone to 534 mg three times a day (1602 mg/day).
DOSAGE FORMS AND STRENGTHS
The 267 mg tablets are yellow, oval shape, film coated tablets debossed with “442" on one side and plain on other side.
The 801 mg tablets are brown, oval shape film coated tablets debossed with “443” on one side and plain on other side.
USE IN SPECIFIC POPULATIONS
- Hepatic Impairment: Monitor for adverse reactions and consider dosage modification or discontinuation of pirfenidone as needed. Pirfenidone is not recommended for use in patients with severe hepatic impairment. (8.6 , 12.3 )
- Renal Impairment: Monitor for adverse reactions and consider dosage modification or discontinuation of pirfenidone as needed. Pirfenidone is not recommended for use in patients with end stage renal disease on dialysis. (8.7 , 12.3 )
- Smokers: Decreased exposure has been noted in smokers which may alter the efficacy profile of pirfenidone. (8.8 )
Pregnancy
Risk Summary
The data with pirfenidone use in pregnant women are insufficient to inform on drug associated risks for major birth defects and miscarriage. In animal reproduction studies, pirfenidone was not teratogenic in rats and rabbits at oral doses up to 3 and 2 times, respectively, the maximum recommended daily dose (MRDD) in adults [see Data ] .
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Data
Animal Data
Animal reproductive studies were conducted in rats and rabbits. In a combined fertility and embryofetal development study, female rats received pirfenidone at oral doses of 0, 50, 150, 450, and 1000 mg/kg/day from 2 weeks prior to mating, during the mating phase, and throughout the periods of early embryonic development from gestation days (GD) 0 to 5 and organogenesis from GD 6 to 17. In an embryofetal development study, pregnant rabbits received pirfenidone at oral doses of 0, 30, 100, and 300 mg/kg/day throughout the period of organogenesis from GD 6 to 18. In these studies, pirfenidone at doses up to 3 and 2 times, respectively, the maximum recommended daily dose (MRDD) in adults (on mg/m 2 basis at maternal oral doses up to 1000 mg/kg/day in rats and 300 mg/kg/day in rabbits, respectively) revealed no evidence of impaired fertility or harm to the fetus due to pirfenidone. In the presence of maternal toxicity, acyclic/irregular cycles (e.g., prolonged estrous cycle) were seen in rats at doses approximately equal to and higher than the MRDD in adults (on a mg/m 2 basis at maternal doses of 450 mg/kg/day and higher). In a pre- and post-natal development study, female rats received pirfenidone at oral doses of 0, 100, 300, and 1000 mg/kg/day from GD 7 to lactation day 20. Prolongation of the gestation period, decreased numbers of live newborn, and reduced pup viability and body weights were seen in rats at an oral dosage approximately 3 times the MRDD in adults (on a mg/m 2 basis at a maternal oral dose of 1000 mg/kg/day).
Lactation
Risk Summary
No information is available on the presence of pirfenidone in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. The lack of clinical data during lactation precludes clear determination of the risk of pirfenidone to an infant during lactation; therefore, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for pirfenidone and the potential adverse effects on the breastfed child from pirfenidone or from the underlying maternal condition.
Data
Animal Data: A study with radio-labeled pirfenidone in rats has shown that pirfenidone or its metabolites are excreted in milk. There are no data on the presence of pirfenidone or its metabolites in human milk, the effects of pirfenidone on the breastfed child, or its effects on milk production.
Pediatric Use
Safety and effectiveness of pirfenidone in pediatric patients have not been established.
Geriatric Use
Of the total number of subjects in the clinical studies receiving pirfenidone, 714 (67%) were 65 years old and over, while 231 (22%) were 75 years old and over. No overall differences in safety or effectiveness were observed between older and younger patients. No dosage adjustment is required based upon age.
Hepatic Impairment
Pirfenidone should be used with caution in patients with mild (Child Pugh Class A) to moderate (Child Pugh Class B) hepatic impairment. Monitor for adverse reactions and consider dosage modification or discontinuation of pirfenidone as needed [see Dosage and Administration (2.3) ] .
The safety, efficacy, and pharmacokinetics of pirfenidone have not been studied in patients with severe hepatic impairment. Pirfenidone is not recommended for use in patients with severe (Child Pugh Class C) hepatic impairment [see Clinical Pharmacology (12.3) ] .
Renal Impairment
Pirfenidone should be used with caution in patients with mild (CLcr 50–80 mL/min), moderate (CLcr 30–50 mL/min), or severe (CLcr less than 30 mL/min) renal impairment [see Clinical Pharmacology (12.3) ]. Monitor for adverse reactions and consider dosage modification or discontinuation of pirfenidone as needed [see Dosage and Administration (2.3) ] . The safety, efficacy, and pharmacokinetics of pirfenidone have not been studied in patients with end-stage renal disease requiring dialysis. Use of pirfenidone in patients with end-stage renal diseases requiring dialysis is not recommended.
Smokers
Smoking causes decreased exposure to pirfenidone [see Clinical Pharmacology (12.3) ], which may alter the efficacy profile of pirfenidone. Instruct patients to stop smoking prior to treatment with pirfenidone and to avoid smoking when using pirfenidone.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Elevated liver enzymes and drug-induced liver injury: ALT, AST, and bilirubin elevations have occurred with pirfenidone including cases of drug-induced liver injury. In the postmarketing setting, non-serious and serious cases of drug-induced liver injury, including severe liver injury with fatal outcomes, have been reported. Monitor ALT, AST, and bilirubin before and during treatment. Temporary dosage reductions or discontinuations may be required. (2.1 , 5.1 )
- Photosensitivity and rash: Photosensitivity and rash have been noted with pirfenidone. Avoid exposure to sunlight and sunlamps. Wear sunscreen and protective clothing daily. Temporary dosage reductions or discontinuations may be required. (5.2 )
- Gastrointestinal disorders: Nausea, vomiting, diarrhea, dyspepsia, gastroesophageal reflux disease, and abdominal pain have occurred with pirfenidone. Temporary dosage reductions or discontinuations may be required. (5.3 )
Elevated Liver Enzymes and Drug-Induced Liver Injury
Cases of drug-induced liver injury (DILI) have been observed with pirfenidone. In the postmarketing period, non-serious and serious cases of DILI, including severe liver injury with fatal outcome, have been reported. Patients treated with pirfenidone 2403 mg/day in three Phase 3 trials had a higher incidence of elevations in ALT or AST ≥3 × ULN than placebo patients (3.7% vs. 0.8%, respectively). Elevations ≥10 × ULN in ALT or AST occurred in 0.3% of patients in the pirfenidone 2403 mg/day group and in 0.2% of patients in the placebo group. Increases in ALT and AST ≥3 × ULN were reversible with dose modification or treatment discontinuation.
Conduct liver function tests (ALT, AST, and bilirubin) prior to the initiation of therapy with pirfenidone, monthly for the first 6 months, every 3 months thereafter, and as clinically indicated. Measure liver function tests promptly in patients who report symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine, or jaundice. Dosage modification or interruption may be necessary for liver enzyme elevations [see Dosage and Administration (2.1 , 2.3) ].
Photosensitivity Reaction or Rash
Patients treated with pirfenidone 2403 mg/day in the three Phase 3 studies had a higher incidence of photosensitivity reactions (9%) compared with patients treated with placebo (1%). The majority of the photosensitivity reactions occurred during the initial 6 months. Instruct patients to avoid or minimize exposure to sunlight (including sunlamps), to use a sunblock (SPF 50 or higher), and to wear clothing that protects against sun exposure. Additionally, instruct patients to avoid concomitant medications known to cause photosensitivity. Dosage reduction or discontinuation may be necessary in some cases of photosensitivity reaction or rash [see Dosage and Administration (2.3) ] .
Gastrointestinal Disorders
In the clinical studies, gastrointestinal events of nausea, diarrhea, dyspepsia, vomiting, gastro-esophageal reflux disease, and abdominal pain were more frequently reported by patients in the pirfenidone treatment groups than in those taking placebo. Dosage reduction or interruption for gastrointestinal events was required in 18.5% of patients in the 2403 mg/day group, as compared to 5.8% of patients in the placebo group; 2.2% of patients in the pirfenidone 2403 mg/day group discontinued treatment due to a gastrointestinal event, as compared to 1.0% in the placebo group. The most common (>2%) gastrointestinal events that led to dosage reduction or interruption were nausea, diarrhea, vomiting, and dyspepsia. The incidence of gastrointestinal events was highest early in the course of treatment (with highest incidence occurring during the initial 3 months) and decreased over time. Dosage modifications may be necessary in some cases of gastrointestinal adverse reactions [see Dosage and Administration (2.3) ] .
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Liver Enzyme Elevations and Drug-Induced Liver Injury [see Warnings and Precautions (5.1) ]
- Photosensitivity Reaction or Rash [see Warnings and Precautions (5.2) ]
- Gastrointestinal Disorders [see Warnings and Precautions (5.3) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of pirfenidone has been evaluated in more than 1400 subjects with over 170 subjects exposed to pirfenidone for more than 5 years in clinical trials.
Pirfenidone was studied in 3 randomized, double-blind, placebo-controlled trials (Studies 1, 2, and 3) in which a total of 623 patients received 2403 mg/day of pirfenidone and 624 patients received placebo. Subjects ages ranged from 40 to 80 years (mean age of 67 years). Most patients were male (74%) and Caucasian (95%). The mean duration of exposure to pirfenidone was 62 weeks (range: 2 to 118 weeks) in these 3 trials.
At the recommended dosage of 2403 mg/day, 14.6% of patients on pirfenidone compared to 9.6% on placebo permanently discontinued treatment because of an adverse event. The most common (>1%) adverse reactions leading to discontinuation were rash and nausea. The most common (>3%) adverse reactions leading to dosage reduction or interruption were rash, nausea, diarrhea, and photosensitivity reaction.
The most common adverse reactions with an incidence of ≥10% and more frequent in the pirfenidone than placebo treatment group are listed in Table 2.
| Adverse Reaction | % of Patients (0 to 118 Weeks) | |
|---|---|---|
| Pirfenidone 2403 mg/day (N = 623) | Placebo (N = 624) | |
| Nausea | 36% | 16% |
| Rash | 30% | 10% |
| Abdominal Pain Includes abdominal pain, upper abdominal pain, abdominal distension, and stomach discomfort. | 24% | 15% |
| Upper Respiratory Tract Infection | 27% | 25% |
| Diarrhea | 26% | 20% |
| Fatigue | 26% | 19% |
| Headache | 22% | 19% |
| Decreased Appetite | 21% | 8% |
| Dyspepsia | 19% | 7% |
| Dizziness | 18% | 11% |
| Vomiting | 13% | 6% |
| Gastro-esophageal Reflux Disease | 11% | 7% |
| Sinusitis | 11% | 10% |
| Insomnia | 10% | 7% |
| Weight Decreased | 10% | 5% |
| Arthralgia | 10% | 7% |
Adverse reactions occurring in ≥5 to <10% of pirfenidone-treated patients and more commonly than placebo are photosensitivity reaction (9% vs. 1%), pruritus (8% vs. 5%), asthenia (6% vs. 4%), dysgeusia (6% vs. 2%), and non-cardiac chest pain (5% vs. 4%).
Postmarketing Experience
In addition to adverse reactions identified from clinical trials the following adverse reactions have been identified during post-approval use of pirfenidone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency.
Blood and Lymphatic System Disorders Agranulocytosis
Immune System Disorders Angioedema
Hepatobiliary Disorders Drug-induced liver injury [see Warnings and Precautions (5.1) ]
DRUG INTERACTIONS
Moderate (e.g., ciprofloxacin) and strong inhibitors of CYP1A2 (e.g., fluvoxamine) increase systemic exposure of pirfenidone and may alter the adverse reaction profile of pirfenidone. Discontinue fluvoxamine prior to administration of pirfenidone or reduce to 267 mg three times a day. Consider dosage reduction with use of ciprofloxacin. (7.1 )
CYP1A2 Inhibitors
Pirfenidone is metabolized primarily (70 to 80%) via CYP1A2 with minor contributions from other CYP isoenzymes including CYP2C9, 2C19, 2D6 and 2E1.
Strong CYP1A2 Inhibitors
The concomitant administration of pirfenidone and fluvoxamine or other strong CYP1A2 inhibitors (e.g., enoxacin) is not recommended because it significantly increases exposure to pirfenidone [see Clinical Pharmacology (12.3) ]. Use of fluvoxamine or other strong CYP1A2 inhibitors should be discontinued prior to administration of pirfenidone and avoided during pirfenidone treatment. In the event that fluvoxamine or other strong CYP1A2 inhibitors are the only drug of choice, dosage reductions are recommended. Monitor for adverse reactions and consider discontinuation of pirfenidone as needed [see Dosage and Administration (2.4) ].
Moderate CYP1A2 Inhibitors
Concomitant administration of pirfenidone and ciprofloxacin (a moderate inhibitor of CYP1A2) moderately increases exposure to pirfenidone [see Clinical Pharmacology (12.3) ]. If ciprofloxacin at the dosage of 750 mg twice daily cannot be avoided, dosage reductions are recommended [see Dosage and Administration (2.4) ]. Monitor patients closely when ciprofloxacin is used at a dosage of 250 mg or 500 mg once daily.
Concomitant CYP1A2 and other CYP Inhibitors
Agents or combinations of agents that are moderate or strong inhibitors of both CYP1A2 and one or more other CYP isoenzymes involved in the metabolism of pirfenidone (i.e., CYP2C9, 2C19, 2D6, and 2E1) should be discontinued prior to and avoided during pirfenidone treatment.
CYP1A2 Inducers
The concomitant use of pirfenidone and a CYP1A2 inducer may decrease the exposure of pirfenidone and this may lead to loss of efficacy. Therefore, discontinue use of strong CYP1A2 inducers prior to pirfenidone treatment and avoid the concomitant use of pirfenidone and a strong CYP1A2 inducer [see Clinical Pharmacology (12.3) ] .
DESCRIPTION
Pirfenidone belongs to the chemical class of pyridone. Pirfenidone is available as film-coated tablets containing 267 mg (yellow) and 801 mg (brown) pirfenidone.
Pirfenidone has a molecular formula of C 12 H 11 NO and a molecular weight of 185.23. Pirfenidone has the following structural formula, which has been referred to as 5-methyl-1phenyl-2-1(H)-pyridone or 5-methyl-1-phenyl-2-(1H)-pyridone.
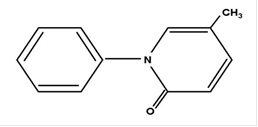
Pirfenidone is a white or pale yellow, crystalline powder. It is sparingly soluble in water, freely soluble in ethanol (96%), very slightly soluble in heptane. The melting point is approximately 109°C.
Pirfenidone tablets contain pirfenidone and the following inactive ingredients: colloidal silicon dioxide, copovidone, croscarmellose sodium, magnesium stearate, silicified microcrystalline cellulose, hypromellose, titanium dioxide, macrogol (polyethylene glycol). Additionally 267 mg tablets contain iron oxide yellow, iron oxide red and 801 mg tablets contain iron oxide yellow, iron oxide red, iron oxide black.
CLINICAL PHARMACOLOGY
Mechanism of Action
The mechanism of action of pirfenidone in the treatment of IPF has not been established.
Pharmacodynamics
Cardiac Electrophysiology:
The effect of pirfenidone on QT interval was evaluated in a randomized, placebo, and positive controlled parallel study in 160 healthy adult volunteers. Volunteers received pirfenidone 2403 mg/day (recommended dose) and 4005 mg/day (1.6 times recommended dose) or placebo for 10 days or a single dose of 400 mg moxifloxacin (active control).
Relative to placebo, the maximum mean change from baseline in study-specific QT interval was 3.2 milliseconds (ms) and 2.2 ms for pirfenidone 2403 mg/day and 4005 mg/day, respectively. No volunteer had a QTc interval greater than 480 ms or change from baseline greater than 60 ms. Although there was no evidence that pirfenidone prolonged the QTc interval in this study, a definitive conclusion may not be drawn as the positive control (moxifloxacin) did not perform as expected in this study, and pirfenidone at 4005 mg/day (1.7 times the maximum recommended dose) did not cover the maximum pirfenidone exposure increase with co-administration of fluvoxamine, a strong CYP1A2 inhibitor.
Pharmacokinetics
Absorption:
After single oral-dose administration of 801 mg pirfenidone (three 267 mg capsules), the maximum observed plasma concentration (C max ) was achieved between 30 minutes and 4 hours (median time of 0.5 hours). Food decreased the rate and extent of absorption. Median T max increased from 0.5 hours to 3 hours with food. Maximum plasma concentrations (C max ) and AUC 0-inf decreased by approximately 49% and 16% with food, respectively.
Bioequivalence was demonstrated in the fasted state when comparing the 801 mg tablet to three 267 mg capsules. The effect of food on pirfenidone exposure was consistent between the tablet and capsule formulations.
A reduced incidence of adverse reactions was observed in the fed group when compared to the fasted group. In controlled studies with IPF patients, pirfenidone was taken with food [see Dosage and Administration (2) and Clinical Studies (14) ].
The absolute bioavailability of pirfenidone has not been determined in humans.
Distribution:
Pirfenidone binds to human plasma proteins, primarily to serum albumin, in a concentration-independent manner over the range of concentrations observed in clinical trials. The overall mean binding was 58% at concentrations observed in clinical studies (1 to 10 mcg/mL). Mean apparent oral volume of distribution is approximately 59 to 71 liters.
Metabolism:
In vitro profiling studies in hepatocytes and liver microsomes have shown that pirfenidone is primarily metabolized in the liver by CYP1A2 and multiple other CYPs (CYP2C9, 2C19, 2D6, and 2E1). Oral administration of pirfenidone results in the formation of four metabolites. In humans, only pirfenidone and 5-carboxy-pirfenidone are present in plasma in significant quantities. The mean metabolite-to-parent ratio ranged from approximately 0.6 to 0.7.
No formal radiolabeled studies have assessed the metabolism of pirfenidone in humans. In vitro data suggests that metabolites are not expected to be pharmacologically active at observed metabolite concentrations.
Elimination:
The mean terminal half-life is approximately 3 hours in healthy subjects. Pirfenidone is excreted predominantly as metabolite 5-carboxy-pirfenidone, mainly in the urine (approximately 80% of the dose). The majority of pirfenidone was excreted as the 5-carboxy metabolite (approximately 99.6% of that recovered).
Specific Populations:
Hepatic Impairment
The pharmacokinetics of pirfenidone and the 5-carboxy-pirfenidone metabolite were studied in 12 subjects with moderate hepatic impairment (Child Pugh Class B) and in 12 subjects with normal hepatic function. Results showed that the mean exposure, AUC 0-inf and C max of pirfenidone increased approximately 1.6- and approximately 1.4-fold in subjects with moderate hepatic impairment, respectively. The exposure of 5-carboxy-pirfenidone did not change significantly in subjects with moderate hepatic impairment.
Renal Impairment
The pharmacokinetics of pirfenidone and the 5-carboxy-pirfenidone metabolite were studied in 18 subjects with mild (CLcr 50 to 80 mL/min), moderate (CLcr 30 to 50 mL/min), and severe (CLcr less than 30 mL/min) renal impairment (n=6/group) and in 6 subjects with normal CLcr (greater than or equal to 80 mL/min) renal function. Results showed that systemic exposure (AUC 0-inf ) to pirfenidone increased approximately 1.4, 1.5, and 1.2-fold in subjects with mild, moderate and severe renal impairment, respectively. The corresponding AUC 0-inf of 5-carboxy-pirfenidone increased 1.7, 3.4, and 5.6-fold, although the change in the patients with mild renal impairment was not statistically significant. The renal clearance of 5-carboxy-pirfenidone decreased significantly in patients with moderate to severe renal impairment.
The pharmacokinetics and safety of pirfenidone has not been studied in subjects with end-stage renal disease requiring dialysis.
Geriatric
Results of population pharmacokinetic analysis suggest that no dosage adjustment is needed in geriatric patients.
Gender
Results of population pharmacokinetic analysis of pirfenidone showed no significant differences in pharmacokinetics between males and females.
Obesity
Results of population pharmacokinetic analysis showed that obesity (Body Mass Index [BMI] greater than or equal to 30 kg/m 2 ) has no significant effect on the pharmacokinetics of pirfenidone.
Race
Population pharmacokinetic analysis showed that race has no significant effect on the pharmacokinetics of pirfenidone.
Drug Interaction Studies:
Cytochrome P450 1A2 Inhibitors
Pirfenidone is a substrate of cytochrome P450 1A2. In a single-dose drug interaction study in 25 healthy nonsmokers and 25 smokers, pirfenidone was coadministered with fluvoxamine (50 mg at bedtime for 3 days; 50 mg twice a day for 3 days, and 50 mg in the morning and 100 mg at bedtime for 4 days). An approximately 4-fold increase in exposure to pirfenidone in nonsmokers and approximately 7-fold increase in exposure in smokers was observed.
In a single-dose drug interaction study in 27 healthy subjects, coadministration of 801 mg of pirfenidone and 750 mg of ciprofloxacin (a moderate inhibitor of CYP1A2) on Day 6 (ciprofloxacin was dosed at 750 mg twice daily from Day 2 to Day 7) increased the exposure to pirfenidone by 81%.
Cytochrome P450 1A2 Inducers
Following a single oral dose of 801 mg pirfenidone in 25 smokers and 25 healthy nonsmokers, the systemic exposure in smokers was significantly lower compared to nonsmokers. AUC 0-inf and C max of pirfenidone in smokers were 46% and 68% of those in nonsmokers, respectively.
Inhibitory Effect of Pirfenidone on P-glycoprotein (Pgp)
The potential for pirfenidone to inhibit Pgp mediated transport of digoxin (5.0 microM) was evaluated in the absence and presence of pirfenidone at concentrations ranging from 1 to 1000 microM in in vitro system. Pirfenidone showed weak inhibition (10 to 30%) of Pgp facilitated digoxin B-A efflux at concentrations of 100 microM and above. Effect of pirfenidone upon Pgp substrate pharmacokinetics and safety has not been evaluated in humans.
Inhibitory Effect of Pirfenidone on CYP2C9, 2C19 or 1A2, 2D6, 3A4
The potential for pirfenidone to inhibit CYP2C9, 2C19 or 1A2 was evaluated in vitro at concentrations up to 1000 microM (approximately 10-fold the mean human C max ). Pirfenidone showed a concentration-dependent inhibition on CYP2C9, 2C19 or 1A2, 2D6, and 3A4. At 1000 microM, pirfenidone inhibits the activity of these enzymes by 30.4%, 27.5%, 34.1%, 21%, and 9.6%, respectively. Effect of pirfenidone upon pharmacokinetics and safety of CYP2C9, 2C19, 1A2, 2D6, and 3A4 substrates has not been evaluated in humans.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term studies were conducted in mice and rats with admixture of pirfenidone to the diet to evaluate its carcinogenic potential.
In a 24-month carcinogenicity study in B6C3F1 mice, pirfenidone caused statistically significant dose-related increases of the combination of hepatocellular adenoma and carcinoma and hepatoblastoma in male mice at doses of 800 mg/kg and above (AUC exposure approximately 0.4 times adult exposure at the MRDD). There were statistically significant dose-related increases of the combination of hepatocellular adenoma and carcinoma in female mice at doses of 2000 mg/kg and above (AUC exposure approximately 0.7 times adult exposure at the MRDD).
In a 24-month carcinogenicity study in Fischer rats, pirfenidone caused statistically significant dose-related increases of the combination of hepatocellular adenoma and carcinoma in male rats at doses of 750 mg/kg and above (AUC exposure approximately 1.9 times adult exposure at the MRDD). There were statistically significant increases of the combination of hepatocellular adenoma and carcinoma and the combination of uterine adenocarcinoma and adenoma at a dose of 1500 mg/kg/day (AUC exposure approximately 3.0 times adult exposure at the MRDD).
The relevance of these tumor findings in rodents to humans is unknown.
Mutagenesis
Pirfenidone was not mutagenic or clastogenic in the following tests: mutagenicity tests in bacteria, a chromosomal aberration test in Chinese hamster lung cells, and a micronucleus test in mice.
Impairment of Fertility
Pirfenidone had no effects on fertility and reproductive performance in rats at dosages up to 1000 mg/kg/day (approximately 3 times the MRDD in adults on a mg/m 2 basis).
CLINICAL STUDIES
The efficacy of pirfenidone was evaluated in patients with IPF in three phase 3, randomized, double-blind, placebo-controlled, multicenter trials (Studies 1, 2, and 3).
Study 1 was a 52-week trial comparing pirfenidone 2403 mg/day (n=278) versus placebo (n=277) in patients with IPF. Study 2 and Study 3 were nearly identical to each other in design, with few exceptions, including an intermediate dose treatment arm in Study 2. Study 2 compared treatment with either pirfenidone 2403 mg/day (n=174) or pirfenidone 1197 mg/day (n=87) to placebo (n=174), while Study 3 compared pirfenidone 2403 mg/day (n=171) to placebo (n=173). Study drug was administered three times daily with food for a minimum of 72 weeks. Patients continued on treatment until the last patient completed 72 weeks of treatment, which included observations to approximately 120 weeks of study treatment. The primary endpoint was the change in percent predicted forced vital capacity (%FVC) from baseline to study end, measured at 52 weeks in Study 1, and at 72 weeks in Studies 2 and 3.
Studies 1, 2 and 3 enrolled adult patients who had a clinical and radiographic diagnosis of IPF (with or without accompanying surgical lung biopsy), without evidence or suspicion of an alternative diagnosis for interstitial lung disease. Eligible patients were to have %FVC greater than or equal to 50% at baseline and a percent predicted diffusing capacity of the lungs for carbon monoxide (%DLCO) greater than or equal to 30% (Study 1) or 35% (Studies 2 and 3) at baseline. In all three trials, over 80% of patients completed study treatment.
A total of 1247 patients with IPF were randomized to receive pirfenidone 2403 mg/day (n=623) or placebo (n=624) in these three trials. Baseline characteristics were generally balanced across treatment groups. The study population ranged from 40 to 80 years of age (mean age 67 years). Most patients were male (74%), white (95%), and current or former smokers (65%). Approximately 93% of patients met criteria for definite IPF on high resolution computed tomography (HRCT). Baseline mean %FVC and %DLCO were 72% and 46%, respectively. Approximately 15% subjects discontinued from each treatment group.
Change from Baseline in Percent Predicted Forced Vital Capacity
In Study 1, the primary efficacy analysis for the change in %FVC from baseline to Week 52 demonstrated a statistically significant treatment effect of pirfenidone 2403 mg/day (n=278) compared with placebo (n=277) using a rank ANCOVA with the lowest rank imputation for missing data due to death. In Study 2, there was a statistically significant difference at Week 72 for the change in %FVC from baseline. In Study 3, there was no statistically significant difference at Week 72 for the change in %FVC from baseline.
Figure 1 presents the cumulative distribution for all cut-offs for the change from baseline in %FVC at Week 52 for Study 1. For all categorical declines in lung function, the proportion of patients declining was lower on pirfenidone than on placebo. Study 2 showed similar results.
Figure 1. Cumulative Distribution of Patients by Change in Percent Predicted FVC from Baseline to Week 52 (Study 1). The Dashed Lines Indicate ≥10% Decline or ≥0% Decline. 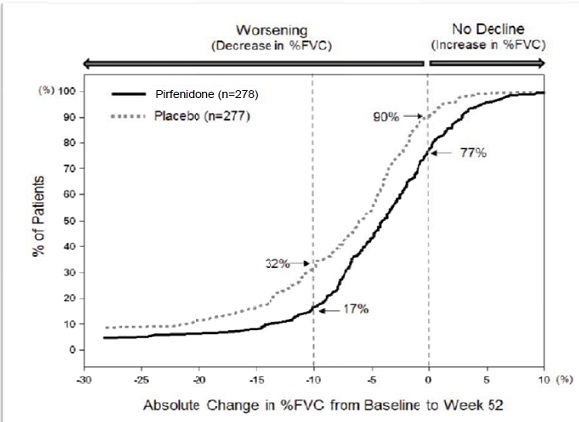
Mean Change from Baseline in FVC (mL)
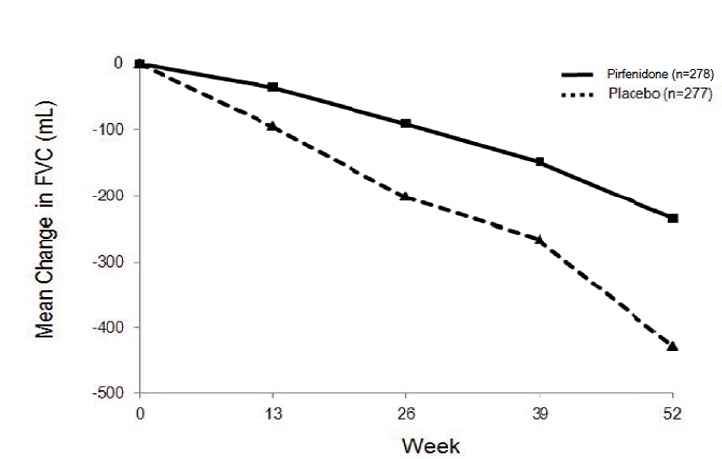
In Study 1, a reduction in the mean decline in FVC (in mL) was observed in patients receiving pirfenidone 2403 mg/day (-235 mL) compared to placebo (-428 mL) (mean treatment difference 193 mL) at Week 52 (see Figure 2 ). In Study 2, a reduction in the decline in FVC volume was also observed in patients receiving pirfenidone 2403 mg/day compared with placebo (mean treatment difference 157 mL) at Week 72. There was no statistically significant difference in decline in FVC volume seen in Study 3.
Figure 2. Mean Change from Baseline in Forced Vital Capacity (Study 1)
Survival
Survival was evaluated for pirfenidone compared to placebo in Studies 1, 2, and 3 as an exploratory analysis to support the primary endpoint (FVC). All-cause mortality was assessed over the study duration and available follow-up period, irrespective of cause of death and whether patients continued treatment. All-cause mortality did not show a statistically significant difference (see Figure3 ).
Figure 3. Kaplan-Meier Estimates of All-Cause Mortality at Vital Status – End of Study: Studies 1, 2, and 3 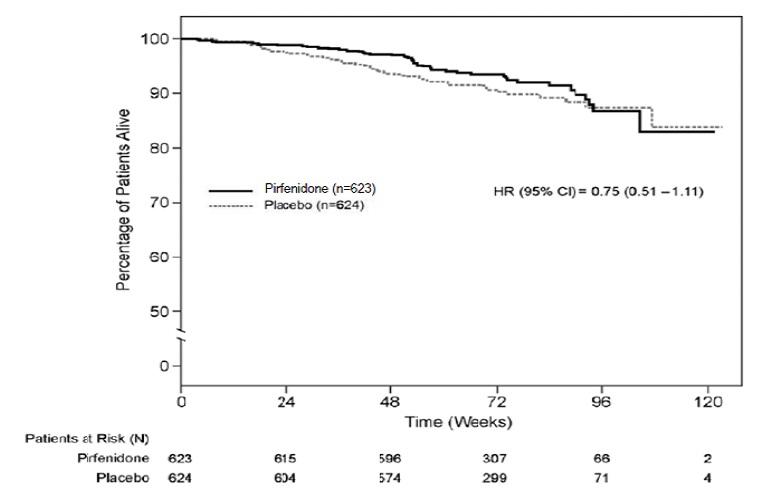
HOW SUPPLIED/STORAGE AND HANDLING
Pirfenidone Tablets, 267 mg are yellow, oval shape, film coated tablets debossed with “442" on one side and plain on other side. They are supplied as follows:
Bottle of 90 Tablets: NDC 76282-717-90
Bottle of 270 Tablets: NDC 76282-717-27
Pirfenidone Tablets, 801 mg are brown, oval shape, film coated tablets debossed with “443" on one side and plain on other side. They are supplied as follows:
Bottle of 90 Tablets: NDC 76282-716-90
Bottle of 270 Tablets: NDC 76282-716-27
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) (see USP Controlled Room Temperature).
Keep the bottle tightly closed. Do not use if the seal over the bottle opening is broken or missing. Safely throw away any pirfenidone that is out of date or no longer needed.
Mechanism of Action
The mechanism of action of pirfenidone in the treatment of IPF has not been established.