Get your patient on Taclonex - Calcipotriene And Betamethasone Dipropionate suspension (Calcipotriene And Betamethasone Dipropionate)
Taclonex - Calcipotriene And Betamethasone Dipropionate suspension prescribing information
INDICATIONS AND USAGE
Taclonex ® Topical Suspension is indicated for the topical treatment of plaque psoriasis of the scalp and body in patients 12 years and older.
DOSAGE AND ADMINISTRATION
Instruct patients to shake bottle prior to using Taclonex Topical Suspension. Apply Taclonex Topical Suspension to affected areas on the scalp and body once daily for up to 8 weeks. Taclonex Topical Suspension should be discontinued when control is achieved. Instruct patients to wash their hands after applying the product. Inform patients that they should not take a bath or shower or wash their hair right after application of Taclonex Topical Suspension.
Patients 12 to 17 years should not use more than 60 grams per week and patients 18 years and older should not use more than 100 grams per week.
Taclonex Topical Suspension should notbe:
- Used with occlusive dressings unless directed by a healthcare provider.
- Used on the face, groin, or axillae, or if skin atrophy is present at the treatment site.
- Applied to the scalp in the 12 hours before or after any chemical treatments to the hair.
Taclonex Topical Suspension is not for oral, ophthalmic, or intravaginal use.
DOSAGE FORMS AND STRENGTHS
Topical Suspension: 0.005%/0.064% - each gram contains 50 mcg of calcipotriene and 0.643 mg of betamethasone dipropionate in a viscous, nearly odorless, almost clear, colorless to slightly off-white suspension.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Available data with Taclonex Topical Suspension are not sufficient to evaluate a drug-associated risk for major birth defects, miscarriages, or adverse maternal or fetal outcomes. Although there are no available data on use of the calcipotriene component in pregnant women, systemic exposure to calcipotriene after topical administration of Taclonex Topical Suspension is likely to be low [see Clinical Pharmacology (12.3) ].
Observational studies suggest an increased risk of having low birth weight infants with the maternal use of potent or super potent topical corticosteroids ( see Data ). Advise pregnant women that Taclonex Topical Suspension may increase the potential risk of having a low birth weight infant and to use Taclonex Topical Suspension on the smallest area of skin and for the shortest duration possible.
In animal reproduction studies, oral administration of calcipotriene to pregnant rats during the period of organogenesis resulted in an increased incidence of minor skeletal abnormalities, including enlarged fontanelles and extra ribs ( see Data ). Oral administration of calcipotriene to pregnant rabbits during the period of organogenesis had no apparent effects on embryo-fetal development. Subcutaneous administration of betamethasone dipropionate to pregnant rats and rabbits during the period of organogenesis resulted in fetal toxicity, including fetal deaths, reduced fetal weight, and fetal malformations (cleft palate and crooked or short tail) ( see Data ). The available data do not allow the calculation of relevant comparisons between the systemic exposures of calcipotriene and betamethasone dipropionate observed in animal studies to the systemic exposures that would be expected in humans after topical use of Taclonex Topical Suspension.
The estimated background risk of major birth defects and miscarriage of the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Available observational studies in pregnant women did not identify a drug-associated risk of major birth defects, preterm delivery, or fetal mortality with the use of topical corticosteroids of any potency. However, when the dispensed amount of potent or super potent topical corticosteroids exceeded 300 grams during the entire pregnancy, maternal use was associated with an increased risk of low birth weight in infants.
Animal Data
Embryo-fetal development studies with calcipotriene were performed by the oral route in rats and rabbits. Pregnant rats received dosages of 0, 6, 18, or 54 mcg/kg/day (0, 36, 108, and 324 mcg/m 2 /day, respectively) on days 6-15 of gestation (the period of organogenesis). There were no apparent effects on maternal survival, behavior, or body weight gain, no effects on litter parameters, and no effects on the incidence of major malformations in fetuses. Fetuses from dams dosed at 54 mcg/kg/day exhibited a significantly increased incidence of minor skeletal abnormalities, including enlarged fontanelles and extra ribs.
Pregnant rabbits were dosed daily with calcipotriene at exposures of 0, 4, 12, or 36 mcg/kg/day (0, 48, 144, and 432 mcg/m 2 /day, respectively) on days 6-18 of gestation (the period of organogenesis). Mean maternal body weight gain was reduced in animals dosed at 12 or 36 mcg/kg/day. The incidence of fetal deaths was increased in the group dosed at 36 mcg/kg/day; reduced fetal weight was also observed in this group. The incidence of major malformations among fetuses was not affected. An increase in the incidence of minor skeletal abnormalities, including incomplete ossification of sternebrae, pubic bones, and forelimb phalanges, was observed in the group dosed at 36 mcg/kg/day.
Embryo-fetal development studies with betamethasone dipropionate were performed via subcutaneous injection in mice and rabbits. Pregnant mice were administered doses of 0, 156, 625, or 2500 mcg/kg/day (0, 468, 1875, and 7500 mcg/m 2 /day, respectively) on days 7 through 13 of gestation (the period of organogenesis). Betamethasone dipropionate induced fetal toxicity, including fetal deaths, reduced fetal weight, malformations (increased incidence of the cleft palate and crooked or short tail), and minor skeletal abnormalities (delayed ossification of vertebra and sternebrae). Fetal toxicity was observed at the lowest exposure that was evaluated (156 mcg/kg/day).
Pregnant rabbits were injected subcutaneously at dosages of 0, 0.625, 2.5, and 10 mcg/kg/day (0, 7.5, 30, and 120 mcg/m 2 /day, respectively) on days 6 through 18 of gestation (the period of organogenesis). Betamethasone dipropionate induced fetal toxicity, including fetal deaths, reduced fetal weight, external malformations (including malformed ears, cleft palate, umbilical hernia, kinked tail, club foot, and club hand), and skeletal malformations (including absence of phalanges of the first digit and cranial dysplasia) at dosages of 2.5 mcg/kg/day and above.
Calcipotriene was evaluated for effects on peri- and post-natal development when orally administered to pregnant rats at dosages of 0, 6, 18 or 54 mcg/kg/day (0, 36, 108, and 324 mcg/m 2 /day, respectively) from gestation day 15 through day 20 postpartum. No remarkable effects were observed on any parameter, including survival, behavior, body weight, litter parameters, or the ability to nurse or rear pups.
Betamethasone dipropionate was evaluated for effects on peri- and post-natal development when orally administered to pregnant rats at dosages of 0, 100, 300, and 1000 mcg/kg/day (0, 600, 1800, and 6000 mcg/m 2 /day, respectively) from gestation day 6 through day 20 postpartum. Mean maternal body weight was significantly reduced on gestation day 20 in animals dosed at 300 and 1000 mcg/kg/day. The mean duration of gestation was slightly, but statistically significantly, increased at 100, 300, and 1000 mcg/kg/day. The mean percentage of pups that survived to day 4 was reduced in relation to dosage. On lactation day 5, the percentage of pups with a reflex to right themselves when placed on their back was significantly reduced at 1000 mcg/kg/day. No effects on the ability of pups to learn were observed, and the ability of the offspring of treated rats to reproduce was not affected.
Lactation
Risk Summary
There is no information regarding the presence of topically administered calcipotriene and betamethasone dipropionate in human milk, the effects on the breastfed infant, or the effects on milk production. Concentrations of calcipotriene in plasma are low after topical administration, and therefore, concentrations in human milk are likely to be low [ see Clinical Pharmacology (12.3) ]. It is not known whether topical administration of large amounts of betamethasone dipropionate could result in sufficient systemic absorption to produce detectable quantities in human milk (see Clinical Considerations ). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Taclonex ® Topical Suspension and any potential adverse effects on the breastfed child from Taclonex Topical Suspension or from the underlying maternal condition.
Clinical Considerations
To minimize potential exposure to the breastfed infant via breast milk, use Taclonex Topical Suspension on the smallest area of skin and for the shortest duration possible while breastfeeding. Advise breastfeeding women not to apply Taclonex Topical Suspension directly to the nipple and areola to avoid direct infant exposure [see Use in Specific Populations (8.4) ] .
Pediatric Use
The safety and effectiveness of Taclonex Topical Suspension for the treatment of plaque psoriasis of the scalp and body have been established in pediatric patients age 12 to 17 years. The use of Taclonex Topical Suspension for this indication is supported by evidence from adequate and well-controlled trials in adults and from three uncontrolled trials in pediatric subjects that enrolled 109 adolescents with moderate psoriasis of the scalp and 107 adolescents with psoriasis of the scalp and body. After 4 weeks of once daily treatment with Taclonex Topical Suspension, HPA axis suppression was observed in 3% of adolescents with psoriasis of the scalp and 16% of adolescents with psoriasis of the scalp and body. Calcium metabolism was evaluated in 107 adolescents with psoriasis of the scalp and body treated with Taclonex Topical Suspension and no cases of hypercalcemia or clinically relevant changes in urinary calcium were reported [see Warnings and Precautions (5.2) , Adverse Reactions (6.1) , and Clinical Pharmacology (12.2) ].
Because of a higher ratio of skin surface area to body mass, pediatric patients are at a greater risk than adults of systemic toxicity when treated with topical corticosteroids. Pediatric patients are, therefore, also at greater risk of HPA axis suppression and adrenal insufficiency with the use of topical corticosteroids including Taclonex ® Topical Suspension [see Clinical Pharmacology (12.2) ].
Rare systemic toxicities such as Cushing's syndrome, linear growth retardation, delayed weight gain, and intracranial hypertension have been reported in pediatric patients, especially those with prolonged exposure to large doses of high potency topical corticosteroids. Local adverse reactions including striae have also been reported with use of topical corticosteroids in pediatric patients.
The safety and effectiveness of Taclonex Topical Suspension in pediatric patients less than 12 years of age have not been established.
Geriatric Use
Clinical studies of Taclonex Topical Suspension in plaque psoriasis on non-scalp areas included 124 subjects who were 65 years of age or older, and 36 were 75 years of age or older. Clinical studies of Taclonex Topical Suspension in subjects with psoriasis of the scalp included 334 subjects who were 65 years or older and 84 subjects who were 75 years or older.
No overall differences in safety or effectiveness of Taclonex Topical Suspension were observed between these subjects and younger subjects, and other reported clinical experience has not identified any differences in responses between the elderly and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Hypercalcemia and Hypercalciuria: Hypercalcemia and hypercalciuria have been reported. If either occurs, discontinue until parameters of calcium metabolism normalize. (5.1 )
- Effects on Endocrine System: Can cause reversible hypothalamic-pituitary-adrenal (HPA) axis suppression with the potential for glucocorticosteroid insufficiency during and after withdrawal of treatment. Risk factors include the use of high-potency topical corticosteroid, use over a large surface area or to areas under occlusion, prolonged use, altered skin barrier, liver failure, and use in pediatric patients. Modify use should HPA axis suppression develop. (5.2 , 8.4 )
- Ophthalmic Adverse Reactions: May increase the risk of cataracts and glaucoma. If visual symptoms occur, consider referral to an ophthalmologist. (5.5 )
Hypercalcemia and Hypercalciuria
Hypercalcemia and hypercalciuria have been observed with use of Taclonex Topical Suspension. If hypercalcemia or hypercalciuria develop, discontinue treatment until parameters of calcium metabolism have normalized. The incidence of hypercalcemia and hypercalciuria following Taclonex Topical Suspension treatment of more than 8 weeks has not been evaluated [see Clinical Pharmacology (12.2) ].
Effects on Endocrine System
Taclonex Topical Suspension can cause reversible hypothalamic-pituitary-adrenal (HPA) axis suppression with the potential for clinical glucocorticosteroid insufficiency. This may occur during treatment or upon withdrawal of treatment. Factors that predispose a patient to HPA axis suppression include the use of high-potency steroids, large treatment surface areas, prolonged use, use of occlusive dressings, altered skin barrier, liver failure, and young age.
Evaluation for HPA axis suppression may be done by using the adrenocorticotropic hormone (ACTH) stimulation test. If HPA axis suppression is documented, gradually withdraw Taclonex Topical Suspension, reduce the frequency of application, or substitute with a less potent corticosteroid.
The following trials evaluated the effects of Taclonex Topical Suspension on HPA axis suppression:
- In a trial evaluating the effects of Taclonex Topical Suspension and Taclonex Ointment on the HPA axis, 32 adult subjects applied both Taclonex Topical Suspension on the scalp and Taclonex Ointment on the body. Adrenal suppression was identified in 5 of 32 subjects (16%) after 4 weeks of treatment and in 2 of 11 subjects (18%) who continued treatment for 8 weeks. In another trial, 36 adult subjects applied Taclonex Topical Suspension on the body and scalp and 7 subjects applied Taclonex Topical Suspension on the body. Adrenal suppression occurred in 3 out of 43 subjects (7%) after 4 weeks of treatment and in none of the 36 subjects who continued treatment for 8 weeks [ see Clinical Pharmacology (12.2) ].
- In two trials, the effects of Taclonex Topical Suspension on the HPA axis were evaluated in 31 and 30 pediatric subjects aged 12 to 17 years old who applied Taclonex Topical suspension on the scalp and the scalp/body, respectively. Adrenal suppression occurred in 1 of 30 evaluable subjects (3%) after 4 weeks of treatment (scalp) and 5 of 31 evaluable subjects (16%) after up to 8 weeks of treatment (scalp and body) [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.2) ].
Cushing's Syndrome and Hyperglycemia
Cushing's syndrome and hyperglycemia may occur due to the systemic effects of the topical corticosteroid. These complications are rare and generally occur after prolonged exposure to excessively large doses, especially of high-potency topical corticosteroids.
Additional Considerations for Endocrine Adverse Reactions
Pediatric patients may be more susceptible to systemic toxicity due to their larger skin surface to body mass ratios [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.2) ].
Use of more than one corticosteroid-containing product at the same time may increase the total systemic corticosteroid exposure.
Allergic Contact Dermatitis with Topical Corticosteroids
Allergic contact dermatitis to a topical corticosteroid is usually diagnosed by observing a failure to heal rather than a clinical exacerbation. Such an observation should be corroborated with appropriate diagnostic patch testing.
Allergic Contact Dermatitis with Topical Calcipotriene
Allergic contact dermatitis has been observed with use of topical calcipotriene. Such an observation should be corroborated with appropriate diagnostic patch testing.
Ophthalmic Adverse Reactions
Use of topical corticosteroids, including Taclonex ® Topical Suspension, may increase the risk of posterior subcapsular cataracts and glaucoma. Cataracts and glaucoma have been reported with the postmarketing use of topical corticosteroid products [see Adverse Reactions (6.2) ]. Avoid contact of Taclonex Topical Suspension with eyes. Taclonex Topical Suspension may cause eye irritation. Advise patients to report any visual symptoms and consider referral to an ophthalmologist for evaluation.
ADVERSE REACTIONS
The most common adverse reactions (≥ 1%) are folliculitis and burning sensation of skin. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact LEO Pharma Inc. at 1-877-494-4536 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials Conducted in Subjects 18 years and older with Psoriasis of the Scalp
The rates of adverse reactions described below were from randomized, multicenter, vehicle- and/or active controlled clinical trials in adult subjects with psoriasis of the scalp [see Clinical Studies (14) ]. Subjects applied study product once daily for 8 weeks, and the median weekly dose was 12.6 grams.
Adverse reactions that occurred in ≥ 1% of subjects treated with Taclonex Topical Suspension and at a rate higher than in subjects treated with vehicle are presented in Table 1.
| Taclonex Topical Suspension | Betamethasone Dipropionate in vehicle | Calcipotriene in vehicle | Vehicle | |
|---|---|---|---|---|
| N=1,953 | N=1,214 | N=979 | N=173 | |
| Event | # of subjects (%) | |||
| Folliculitis | 16 (1%) | 12 (1%) | 5 (1%) | 0 (0%) |
| Burning sensation of skin | 13 (1%) | 10 (1%) | 29 (3%) | 0 (0%) |
Other less common adverse reactions (<1% but >0.1%) were, in decreasing order of incidence: acne, exacerbation of psoriasis, eye irritation, and pustular rash.
In a 52-week trial, adverse reactions that were reported by >1% of subjects treated with Taclonex Topical Suspension were pruritus (3.6%), psoriasis (2.4%), erythema (2.1%), skin irritation (1.4%), and folliculitis (1.2%).
Clinical Trials Conducted in Subjects 18 years and older with Psoriasis of the Body
In randomized, multicenter, vehicle- and/or active controlled clinical trials in adult subjects with plaque psoriasis on non-scalp areas, 824 subjects applied Taclonex Topical Suspension once daily for 8 weeks [see Clinical Studies (14) ]. The median weekly dose was 22.6 grams.
There were no adverse reactions that occurred in ≥1% of subjects treated with Taclonex Topical Suspension and at a rate higher than in subjects treated with vehicle. Other less common adverse reactions (<1% but >0.1%) were, in decreasing order of incidence: rash and folliculitis.
Clinical Trials Conducted in Subjects 12 to 17 years with Psoriasis of the Scalp
In two uncontrolled clinical trials, 109 subjects aged 12 to 17 years with plaque psoriasis of the scalp applied Taclonex ® Topical Suspension once daily for up to 8 weeks. The median weekly dose was 40 grams. Adverse reactions included acne, acneiform dermatitis and application site pruritus (0.9% each) [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.2) ] .
Clinical Trial Conducted in Subjects 12 to 17 years with Psoriasis of the Scalp and Body
In an uncontrolled clinical trial, 107 subjects aged 12 to 17 years with plaque psoriasis of the scalp and body applied Taclonex Topical Suspension once daily for up to 8 weeks. The median weekly dose was 26.6 grams. Adverse reactions were folliculitis, acne, and erythema (0.9% each) [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.2) ] .
Postmarketing Experience
Because adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Postmarketing reports for local adverse reactions to topical corticosteroids included atrophy, striae, telangiectasias, itching, dryness, hypopigmentation, perioral dermatitis, secondary infection, and miliaria.
Ophthalmic adverse reactions of cataracts, glaucoma, increased intraocular pressure, and central serous chorioretinopathy have been reported during use of topical corticosteroids, including topical betamethasone products.
DESCRIPTION
Taclonex Topical Suspension contains calcipotriene hydrate and betamethasone dipropionate. It is for topical use only. Calcipotriene hydrate is a synthetic vitamin D 3 analog.
Calcipotriene Hydrate
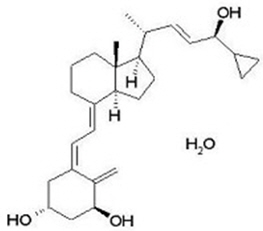
Calcipotriene hydrate is a vitamin D analog and has the chemical name 9,10-secochola-5,7,10(19),22-tetraene-1,3,24-triol,24-cyclo-propyl-,monohydrate, (1α,3β,5Z,7E,22E,24S) with the empirical formula C 27 H 40 O 3 ∙H 2 O, a molecular weight of 430.6, and the following structural formula (calcipotriene hydrate is a white to almost white, crystalline compound):
Betamethasone Dipropionate
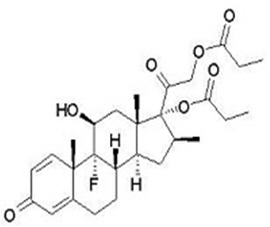
Betamethasone dipropionate is a synthetic corticosteroid and has the chemical name pregna-1,4-diene-3,20-dione-9-fluoro-11-hydroxy-16-methyl-17,21- bis (1-oxypropoxy)-(11β,16β), with the empirical formula C 28 H 37 FO 7 , a molecular weight of 504.6, and the following structural formula (betamethasone dipropionate is a white to almost white, crystalline powder):
Taclonex ® Topical Suspension
Each gram of Taclonex Topical Suspension contains 50 mcg of calcipotriene (equivalent to 52.2 mcg of calcipotriene hydrate) and 0.643 mg of betamethasone dipropionate (equivalent to 0.5 mg of betamethasone) in a base of hydrogenated castor oil, polyoxypropylene stearyl ether, all- rac -alpha-tocopherol, butylhydroxytoluene, and mineral oil. Taclonex Topical Suspension is an odorless, clear to slightly off-white suspension.
CLINICAL PHARMACOLOGY
Mechanism of Action
Taclonex Topical Suspension combines the pharmacological effects of calcipotriene hydrate as a synthetic vitamin D 3 analog and betamethasone dipropionate as a synthetic corticosteroid. However, while their pharmacologic and clinical effects are known, the exact mechanisms of their actions in the treatment of plaque psoriasis of the scalp and body are unknown.
Pharmacodynamics
Hypothalamic-Pituitary-Adrenal (HPA) Axis Suppression:
HPA axis suppression was evaluated in four trials (Trial A, B, C, and D) following the application of Taclonex Topical Suspension. In all these trials, adrenal suppression was defined by a 30-minute post-stimulation cortisol level ≤18 mcg/dL.
- In Trial A, HPA axis suppression was evaluated in adult subjects (N=32) with extensive psoriasis involving at least 30% of the scalp and, in total, 15-30% of the body surface area. Treatment consisted of once daily application of Taclonex Topical Suspension on the scalp in combination with Taclonex Ointment on the body for 4 to 8 weeks. Adrenal suppression was observed in 5 of 32 subjects (16%) after 4 weeks of treatment and in 2 of 11 subjects (18%) who continued treatment for 8 weeks.
- In Trial B, HPA axis suppression was evaluated in adult subjects (N=43) with extensive psoriasis involving 15-30% of the body surface area (including the scalp). Treatment consisted of once daily application of Taclonex Topical Suspension to the body (including the scalp in 36 out of 43 subjects) for 4 to 8 weeks. Adrenal suppression was observed in 3 out of 43 subjects (7%) after 4 weeks of treatment and in none of the 36 subjects (0%) who continued treatment for 8 weeks.
- In Trial C, HPA axis suppression was evaluated in pediatric subjects 12 to 17 years (N=30) with plaque psoriasis of the scalp involving at least 20% of the scalp area. Treatment consisted of once daily application of Taclonex Topical Suspension to the affected area on the scalp for up to 8 weeks. Adrenal suppression was observed in 1 of 30 evaluable subjects (3%) after 4 weeks of treatment and in no subjects (0%) who continued treatment for 8 weeks [see Use in Specific Populations (8.4) ] .
- In Trial D, HPA axis suppression was evaluated in a subset of pediatric subjects aged 12 to 17 years (N=31) with plaque psoriasis of the scalp and body involving 10% to 29% of the body surface area. Adrenal suppression was observed in 5 of 31 subjects (16%): 3 subjects after 4 weeks of treatment, 1 subject after 8 weeks of treatment, and 1 subject after both 4 and 8 weeks of treatment [see Use in Specific Populations (8.4) ] .
Effects on Calcium Metabolism
The effect on calcium metabolism was evaluated following the application of Taclonex Topical Suspension (these trials are described above).
- In Trial A, elevated urinary calcium levels outside the normal range were observed in two subjects (one at 4 weeks and one at 8 weeks).
- In Trial B, there was no change in mean serum or urinary calcium levels. Elevated urinary calcium levels outside the normal range were observed in two subjects (one at 4 weeks and one at 8 weeks).
- In Trial C (N=109), including 31 subjects with at least 20% scalp involvement and 78 subjects with at least 10% scalp involvement, no cases of hypercalcemia and no clinically relevant changes in urinary calcium were reported.
- In Trial D (N=107), no cases of hypercalcemia and no clinically relevant changes in urinary calcium were reported.
Pharmacokinetics
Absorption
The systemic effect of Taclonex Topical Suspension in psoriasis was investigated in Trials A, B, and D [see Clinical Pharmacology (12.2) ].
In Trial A, the serum levels of calcipotriene and betamethasone dipropionate and their major metabolites were measured after 4 and 8 weeks of once daily application of Taclonex ® Topical Suspension on the scalp in combination with Taclonex Ointment on the body. Calcipotriene and betamethasone dipropionate were below the lower limit of quantification in all serum samples of the 34 subjects evaluated. However, one major metabolite of calcipotriene (MC1080) was quantifiable in 10 of 34 (29%) subjects at week 4 and in 5 of 12 (42%) subjects at week 8. The major metabolite of betamethasone dipropionate, betamethasone 17-propionate (B17P) was also quantifiable in 19 of 34 (56%) subjects at week 4 and 7 of 12 (58%) subjects at week 8. The serum concentrations for MC1080 ranged from 20-75 pg/mL. The clinical significance of this finding is unknown.
In Trial B, the plasma levels of calcipotriene and betamethasone dipropionate and their major metabolites were measured after 4 weeks of once daily application of Taclonex Topical Suspension. Calcipotriene and its metabolite MC1080 were below the lower limit of quantification in all plasma samples. Betamethasone dipropionate was quantifiable in 4 of 43 (9) subjects. The metabolite of betamethasone dipropionate (B17P) was quantifiable in 16 of 43 (37%) subjects. The plasma concentrations of betamethasone dipropionate ranged from 30.9-63.5 pg/mL and that of its metabolite betamethasone 17-propionate ranged from 30.5-257 pg/mL. The clinical significance of this finding is unknown.
In Trial D, the plasma levels of calcipotriene and betamethasone dipropionate and their major metabolites were measured after 4 weeks of once daily application of Taclonex Topical Suspension. Calcipotriene and its metabolite MC1080 were below the lower limit of quantification in all plasma samples. Betamethasone dipropionate was quantifiable in 4 of 32 (13%) subjects. The metabolite of betamethasone dipropionate (B17P) was quantifiable in 5 of 32 (16%) subjects. The plasma concentrations of betamethasone dipropionate ranged from 41.4-104 pg/mL and that of its metabolite betamethasone 17-propionate ranged from 30.1-126 pg/mL. The clinical significance of this finding is unknown.
Elimination
Metabolism
Calcipotriene: Calcipotriene metabolism following systemic uptake is rapid and occurs in the liver. The primary metabolites of calcipotriene are less potent than the parent compound.
Calcipotriene is metabolized to MC1046 (the α,β-unsaturated ketone analog of calcipotriene), which is metabolized further to MC1080 (a saturated ketone analog). MC1080 is the major metabolite in plasma. MC1080 is slowly metabolized to calcitroic acid.
Betamethasone dipropionate : Betamethasone dipropionate is metabolized to betamethasone 17-propionate and betamethasone, including the 6β-hydroxy derivatives of those compounds by hydrolysis. Betamethasone 17-propionate (B17P) is the primary metabolite.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
When calcipotriene was applied topically to mice for up to 24 months at dosages of 3, 10, and 30 mcg/kg/day (9, 30, and 90 mcg/m 2 /day, respectively), no significant changes in tumor incidence were observed when compared to control.
A 104-week oral carcinogenicity study was conducted with calcipotriene in male and female rats at doses of 1, 5 and 15 mcg/kg/day (6, 30, and 90 mcg/m 2 /day, respectively). Beginning week 71, the dosage for high-dose animals of both genders was reduced to 10 mcg/kg/day (60 mcg/m 2 /day). A treatment-related increase in benign C-cell adenomas was observed in the thyroid of females that received 15 mcg/kg/day. A treatment-related increase in benign pheochromocytomas was observed in the adrenal glands of males that received 15 mcg/kg/day. No other statistically significant differences in tumor incidence were observed when compared to control. The relevance of these findings to patients is unknown.
When betamethasone dipropionate was applied topically to CD-1 mice for up to 24 months at dosages approximating 1.3, 4.2, and 8.5 mcg/kg/day in females, and 1.3, 4.2, and 12.9 mcg/kg/day in males (up to 26 mcg/m 2 /day and 39 mcg/m 2 /day, in females and males, respectively), no significant changes in tumor incidence were observed when compared to control.
When betamethasone dipropionate was administered via oral gavage to male and female Sprague Dawley rats for up to 24 months at dosages of 20, 60, and 200 mcg/kg/day (120, 360, and 1200 mcg/m 2 /day, respectively), no significant changes in tumor incidence were observed when compared to control.
Calcipotriene did not elicit any genotoxic effects in the Ames mutagenicity assay, the mouse lymphoma TK locus assay, the human lymphocyte chromosome aberration test, or the mouse micronucleus test. Betamethasone dipropionate did not elicit any genotoxic effects in the Ames mutagenicity assay, the mouse lymphoma TK locus assay, or in the rat micronucleus test.
Studies in rats with oral doses of up to 54 mcg/kg/day (324 mcg/m 2 /day) of calcipotriene indicated no impairment of fertility or general reproductive performance. Studies in male rats at oral doses of up to 200 mcg/kg/day (1200 mcg/m 2 /day), and in female rats at oral doses of up to 1000 mcg/kg/day (6000 mcg/m 2 /day), of betamethasone dipropionate indicated no impairment of fertility.
CLINICAL STUDIES
Clinical Trials Conducted in Subjects 18 Years and Older with Psoriasis of the Scalp
Two multicenter, randomized, double-blind trials were conducted in adult subjects with moderate to very severe psoriasis of the scalp.
- In Trial One, 1,407 subjects were randomized to 1 of 4 treatment groups: Taclonex ® Topical Suspension, betamethasone dipropionate in the same vehicle, calcipotriene in the same vehicle, or the vehicle alone.
- In Trial Two, 1,280 subjects were randomized to 1 of 3 treatment groups: Taclonex Topical Suspension, betamethasone dipropionate in the same vehicle, or calcipotriene in the same vehicle.
Both trials enrolled subjects with moderate to very severe psoriasis of the scalp. The majority of subjects had disease of moderate severity at baseline. Subjects were treated once daily for 8 weeks. Efficacy was assessed as the proportion of subjects at Week 8 with absent or very mild disease according to the Investigator's Global Assessment of Disease Severity. "Clear" was defined as no evidence of redness, thickness or scaling. "Almost clear" was defined as an overall clinical picture of lesions with the presence of minimal erythema. Table 2 contains the response rates in each of these 2 trials.
| Taclonex Topical Suspension | Betamethasone Dipropionate in vehicle | Calcipotriene in vehicle | Vehicle | |
|---|---|---|---|---|
| Trial One | (N=494) | (N=531) | (N=256) | (N=126) |
| Week 2 | 55.5% | 46.1% | 18.4% | 9.5% |
| Week 8 | 70.0% | 63.1% | 36.7% | 19.8% |
| Trial Two | (N=512) | (N=517) | (N=251) | - |
| Week 2 | 47.1% | 36.4% | 12.7% | - |
| Week 8 | 67.2% | 59.6% | 41.0% | - |
Clinical Trials Conducted in Subjects 18 Years and Older with Psoriasis of the Body
One multicenter, randomized, double-blind trial was conducted in subjects with mild to moderate plaque psoriasis on non-scalp areas, excluding face, axillae, and groin. In this trial, 1152 subjects were randomized to 1 of 4 treatment groups: Taclonex Topical Suspension, betamethasone dipropionate in the same vehicle, calcipotriene in the same vehicle, or the vehicle alone. Seventy eight percent (78%) of subjects had disease of moderate severity at baseline. Subjects were treated once daily for 8 weeks. Efficacy was assessed at Week 4 and Week 8 as the proportion of subjects who were "Clear" or "Almost clear" according to the Investigator's Global Assessment of Disease Severity. Subjects with mild disease at baseline were required to be "Clear" to be considered a success. Table 3 contains the response rates in this trial.
| Taclonex Topical Suspension | Betamethasone Dipropionate in vehicle | Calcipotriene in vehicle | Vehicle | |
|---|---|---|---|---|
| (N=482) | (N=479) | (N=96) | (N=95) | |
| Week 4 | 13.3% | 12.5% | 5.2% | 2.1% |
| Week 8 | 29.0% | 21.5% | 14.6% | 6.3% |
HOW SUPPLIED/STORAGE AND HANDLING
Taclonex (calcipotriene and betamethasone dipropionate) Topical Suspension, 0.005%/0.064% is a viscous, nearly odorless, almost clear, colorless to slightly off-white suspension. It is available as:
- 60 gram bottle (NDC 50222-501-06)
- 120 gram (2 bottles of 60 gram) (NDC 50222-501-66)
Store between 20°C - 25°C (68°F - 77°F); excursions permitted between 15°C - 30°C (59°F - 86°F). [See USP controlled room temperature]. Do not refrigerate.
Keep the bottle in the carton when not in use. Unused product should be discarded six months after the bottle has been opened. Shake before use. Keep out of reach of children.
INSTRUCTIONS FOR USE
TACLONEX ® (TAK-lo-NEKS) (calcipotriene and betamethasone dipropionate) Topical Suspension
Important: Taclonex Topical Suspension is for use on skin only (topical). Do not get Taclonex Topical Suspension near or in your mouth, eyes, or vagina. Read this Instructions for Use before you start using Taclonex Topical Suspension and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
How to apply Taclonex Topical Suspension to your body: Follow your healthcare provider's instructions of how much Taclonex Topical Suspension to use and where to use it. Apply Taclonex Topical Suspension directly to areas affected by plaque psoriasis and gently rub in. Wash your hands after applying Taclonex Topical Suspension, unless you are treating areas on your hands.
How to apply Taclonex Topical Suspension to your scalp: You do not need to wash your hair before you apply Taclonex Topical Suspension.
| Step 1: Shake the bottle before use. Remove the cap from the bottle. (See Figure A ). |
 | Step 2: Locate the area to treat using your fingers and part your hair. (See Figure B ). |
 | Step 3: Squeeze a drop of Taclonex Topical Suspension to your fingertip. (See Figure C ). |
 | Step 4: Use your fingers to apply the drop of Taclonex Topical Suspension directly to scalp affected by plaque psoriasis. Gently rub in. (See Figure D ). |
| Step 5: After applying Taclonex Topical Suspension, put the cap back on the bottle. Step 6: Wash your hands after applying Taclonex Topical Suspension. Do not wash your hair right after you apply Taclonex Topical Suspension to your scalp. | |

How should I store Taclonex ® Topical Suspension?
- Store the Taclonex Topical Suspension at room temperature between 68°F to 77°F (20°C to 25°C).
- Do not refrigerate Taclonex Topical Suspension.
- Keep bottle in the carton when not in use.
- Discard unused Taclonex Topical Suspension 6 months after it has been opened.
Keep Taclonex Topical Suspension and all medicines out of reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by: LEO Laboratories Ltd. 285 Cashel Road Dublin 12, Ireland
Distributed by: LEO Pharma Inc. Madison, NJ 07940, USA
Revised: 7/2019
Mechanism of Action
Taclonex Topical Suspension combines the pharmacological effects of calcipotriene hydrate as a synthetic vitamin D 3 analog and betamethasone dipropionate as a synthetic corticosteroid. However, while their pharmacologic and clinical effects are known, the exact mechanisms of their actions in the treatment of plaque psoriasis of the scalp and body are unknown.