Get your patient on Teriflunomide - Teriflunomide tablet, Coated (Teriflunomide)
Teriflunomide - Teriflunomide tablet, Coated prescribing information
WARNING: HEPATOTOXICITY and EMBRYOFETAL TOXICITY
- Hepatotoxicity Clinically significant and potentially life-threatening liver injury, including acute liver failure requiring transplant, has been reported in patients treated with teriflunomide tablets in the postmarketing setting [see Warnings and Precautions (5.1) ] . Concomitant use of teriflunomide tablets with other hepatotoxic drugs may increase the risk of severe liver injury. Obtain transaminase and bilirubin levels within 6 months before initiation of teriflunomide tablets therapy. Monitor ALT levels at least monthly for six months after starting teriflunomide tablets [see Warnings and Precautions (5.1) ] . If drug induced liver injury is suspected, discontinue teriflunomide tablets and start an accelerated elimination procedure with cholestyramine or charcoal [see Warnings and Precautions (5.3 )] . Teriflunomide tablet is contraindicated in patients with severe hepatic impairment [see Contraindications (4) ]. Patients with pre-existing liver disease may be at increased risk of developing elevated serum transaminases when taking teriflunomide tablets.
- Embryofetal Toxicity Teriflunomide tablet is contraindicated for use in pregnant women and in females of reproductive potential who are not using effective contraception because of the potential for fetal harm. Teratogenicity and embryolethality occurred in animals at plasma teriflunomide exposures lower than that in humans. Exclude pregnancy before the start of treatment with teriflunomide tablets in females of reproductive potential. Advise females of reproductive potential to use effective contraception during teriflunomide tablet treatment and during an accelerated drug elimination procedure after teriflunomide tablet treatment. Stop teriflunomide tablets and use an accelerated drug elimination procedure if the patient becomes pregnant [see Contraindications (4 ), Warnings and Precautions (5.2 , 5.3 ), Use in Specific Populations (8.1 , 8.3 ), and Clinical Pharmacology (12.3 )]
INDICATIONS AND USAGE
Teriflunomide tablet is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
DOSAGE AND ADMINISTRATION
The recommended dose of teriflunomide tablets is 7mg or 14 mg orally once daily. Teriflunomide tablets can be taken with or without food. Monitoring to Assess Safety.
- Obtain transaminase and bilirubin levels within 6 months before initiation of teriflunomide tablet therapy. Monitor ALT levels at least monthly for six months after starting teriflunomide tablets [see Warnings and Precautions (5.1) ] .
- Obtain a complete blood cell count (CBC) within 6 months before the initiation of treatment with teriflunomide tablets. Further monitoring should be based on signs and symptoms of infection [see Warnings and Precautions (5.4) ] .
- Prior to initiating teriflunomide tablets, screen patients for latent tuberculosis infection with a tuberculin skin test or blood test for mycobacterium tuberculosis infection [ see Warnings and Precautions (5.4) ] .
- Exclude pregnancy prior to initiation of treatment with teriflunomide tablets in females of reproductive potential [see Warnings and Precautions (5.2) ].
- Check blood pressure before start of teriflunomide tablet treatment and periodically thereafter [see Warnings and Precautions (5.9 )].
DOSAGE FORMS AND STRENGTHS
Teriflunomide tablets are available as 7 mg and 14 mg tablets.
The 14 mg tablet is a blue (pale blue to pastel blue) colored, round, biconvex, film-coated tablet debossed with the dose strength “14” on one side. Each tablet contains 14 mg of teriflunomide. The 7 mg tablet is a green (light greenish-blue grey to pale greenish-blue) colored, round, biconvex, film-coated tablet debossed with the dose strength “7” on one side. Each tablet contains 7 mg of teriflunomide.
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Teriflunomide during pregnancy. Healthcare providers and patients are encouraged to report pregnancies by calling 1-800-745-4447, option 2.
Risk Summary Teriflunomide tablet is contraindicated for use in pregnant women and females of reproductive potential not using effective contraception because of the potential for fetal harm based on animal data [see Contraindications (4 ) and Warnings and Precautions (5.2 )] .
In animal reproduction studies in rat and rabbit, oral administration of teriflunomide during organogenesis caused teratogenicity and embryolethality at plasma exposures (AUC) lower than that at the maximum recommended human dose (MRHD) of 14 mg/day [see Data ] . Available human data from pregnancy registries, clinical trials, pharmacovigilance cases, and published literature are too limited to draw any conclusions, but they do not clearly indicate increased birth defects or miscarriage associated with inadvertent teriflunomide exposure in the early first trimester when followed by an accelerated elimination procedure [see Clinical Considerations and Data ] . There are no human data pertaining to exposures later in the first trimester or beyond.
In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage in the indicated population is unknown.
Clinical Considerations Fetal/Neonatal adverse reactions
Lowering the plasma concentration of teriflunomide by instituting an accelerated drug elimination procedure as soon as pregnancy is detected may decrease the risk to the fetus from teriflunomide tablets. The accelerated drug elimination procedure includes verification that the plasma teriflunomide concentration is less than 0.02 mg/L [see Warnings and Precautions (5.3 ) and Clinical Pharmacology (12.3 )].
Data Human data
Available human data are limited. Prospectively reported data (from clinical trials and postmarketing reports) from > 150 pregnancies in patients treated with teriflunomide tablets and > 300 pregnancies in patients treated with leflunomide have not demonstrated an increased rate of congenital malformations or miscarriage following teriflunomide exposure in the early first trimester when followed by an accelerated elimination procedure. Specific patterns of major congenital malformations in humans have not been observed. Limitations of these data include an inadequate number of reported pregnancies from which to draw conclusions, the short duration of drug exposure in reported pregnancies, which precludes a full evaluation of the fetal risks, incomplete reporting, and the inability to control for confounders (such as underlying maternal disease and use of concomitant medications).
Animal data
When teriflunomide (oral doses of 1, 3, or 10 mg/kg/day) was administered to pregnant rats throughout the period of organogenesis, high incidences of fetal malformation (primarily craniofacial, and axial and appendicular skeletal defects) and fetal death were observed at doses not associated with maternal toxicity. Adverse effects on fetal development were observed following dosing at various stages throughout organogenesis. Maternal plasma exposure at the no-effect level (1.0 mg/kg/day) for fetal developmental toxicity in rats was less than that in humans at the maximum recommended human dose (MRHD, 14 mg/day).
Administration of teriflunomide (oral doses of 1, 3.5, or 12 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in high incidences of fetal malformation (primarily craniofacial, and axial and appendicular skeletal defects) and fetal death at doses associated with minimal maternal toxicity. Maternal plasma exposure at the no-effect dose (1.0 mg/kg/day) for fetal developmental toxicity in rabbits was less than that in humans at the MRHD.
In studies in which teriflunomide (oral doses of 0.05, 0.1, 0.3, 0.6, or 1.0 mg/kg/day) was administered to rats during gestation and lactation, decreased growth, eye and skin abnormalities, and high incidences of malformation (limb defects) and postnatal death were observed in the offspring at doses not associated with maternal toxicity. Maternal plasma exposure at the no-effect dose for prenatal and postnatal developmental toxicity in rats (0.10 mg/kg/day) was less than that in humans at the MRHD.
In animal reproduction studies of leflunomide, embryolethality and teratogenic effects were observed in pregnant rat and rabbit at or below clinically relevant plasma teriflunomide exposures (AUC). In published reproduction studies in pregnant mice, leflunomide was embryolethal and increased the incidence of malformations (craniofacial, axial skeletal, heart and great vessel). Supplementation with exogenous uridine reduced the teratogenic effects in pregnant mice, suggesting that the mode of action (inhibition of mitochondrial enzyme dihydroorotate dehydrogenase) is the same for therapeutic efficacy and developmental toxicity.
At recommended doses in humans, teriflunomide and leflunomide result in a similar range of plasma concentrations of teriflunomide.
Lactation
Risk Summary There are no data on the presence of teriflunomide in human milk, the effects on the breastfed infant, or the effects on milk production. Teriflunomide was detected in rat milk following a single oral dose. Because of the potential for adverse reactions in a breastfed infant from teriflunomide, women should not breastfeed during treatment with teriflunomide tablets.
Females and Males of Reproductive Potential
Pregnancy Testing Exclude pregnancy prior to initiation of treatment with teriflunomide tablets in females of reproductive potential. Advise females to notify their healthcare provider immediately if pregnancy occurs or is suspected during treatment [see Warnings and Precautions (5.2 , 5.3 ) and Use in Specific Populations (8.1 )].
Contraception Females
Females of reproductive potential should use effective contraception while taking teriflunomide tablets. If teriflunomide tablet is discontinued, use of contraception should be continued until it is verified that plasma concentrations of teriflunomide are less than 0.02 mg/L (0.02 mcg/mL, the level expected to have minimal fetal risk, based on animal data).
Females of reproductive potential who wish to become pregnant should discontinue teriflunomide tablets and undergo an accelerated elimination procedure. Effective contraception should be used until it is verified that plasma concentrations of teriflunomide are less than 0.02 mg/L (0.02 mcg/mL) [see Warnings and Precautions (5.2 , 5.3 ) and Use in Specific Populations (8.1 )] .
Males
Teriflunomide is detected in human semen. Animal studies to specifically evaluate the risk of male mediated fetal toxicity have not been conducted. To minimize any possible risk, men not wishing to father a child and their female partners should use effective contraception. Men wishing to father a child should discontinue use of teriflunomide tablets and either undergo an accelerated elimination procedure or wait until verification that the plasma teriflunomide concentration is less than 0.02 mg/L (0.02 mcg/mL) [see Warnings and Precautions (5.3 )] .
Pediatric Use
Safety and effectiveness in pediatric patients have not been established. Pancreatitis has been reported in adults in the postmarketing setting, but appears to occur at higher frequency in the pediatric population [see Warnings and Precautions (5.11 ) ] . Additionally, elevated or abnormal blood creatine phosphokinase was reported.
Juvenile Animal Toxicity Data Oral administration of teriflunomide (0, 0.3, 3, or 6 mg/kg/day) to young rats on postnatal days 21 to 70 resulted in suppression of immune function (T-cell dependent antibody response) at the mid and high doses, and adverse effects on male reproductive organs (reduced sperm count) and altered neurobehavioral function (increased locomotor activity) at the high dose.
Pediatric information describing a clinical study in which efficacy was not demonstrated is approved for Sanofi-Aventis U.S. LLC’s Aubagio ® (teriflunomide) tablets. However, due to Sanofi-Aventis U.S. LLC’s marketing exclusivity rights, this drug product is not labeled with that information.
Geriatric Use
Clinical studies of teriflunomide tablets did not include patients over 65 years old.
Hepatic Impairment
No dosage adjustment is necessary for patients with mild and moderate hepatic impairment. The pharmacokinetics of teriflunomide in severe hepatic impairment has not been evaluated.
Teriflunomide tablet is contraindicated in patients with severe hepatic impairment [see Contraindications (4 ), Warnings and Precautions (5.1 ), and Clinical Pharmacology (12.3 )] .
Renal Impairment
No dosage adjustment is necessary for patients with mild, moderate, and severe renal impairment [see Clinical Pharmacology (12.3 )] .
CONTRAINDICATIONS
Teriflunomide tablet is contraindicated in/with:
- Patients with severe hepatic impairment [see Warnings and Precautions (5.1) ] .
- Pregnant women and females of reproductive potential not using effective contraception. Teriflunomide tablets may cause fetal harm [see Warnings and Precautions (5.2 , 5.3) and Use in Specific Populations (8.1) ].
- Patients with a history of a hypersensitivity reaction to teriflunomide, leflunomide, or to any of the inactive ingredients in teriflunomide tablets. Reactions have included anaphylaxis, angioedema, and serious skin reactions [see Warnings and Precautions (5.5) ].
- Co-administration with leflunomide [see Clinical Pharmacology (12.3) ].
WARNINGS AND PRECAUTIONS
- Elimination of teriflunomide can be accelerated by administration of cholestyramine or activated charcoal for 11 days. (5.3 )
- Teriflunomide tablets may decrease WBC. A recent CBC should be available before starting teriflunomide tablets. Monitor for signs and symptoms of infection. Consider suspending treatment with teriflunomide tablets in case of serious infection. Do not start teriflunomide tablets in patients with active infections. (5.4 )
- Stop teriflunomide tablets if patient has anaphylaxis, angioedema, Stevens-Johnson syndrome, toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms; initiate rapid elimination. (5.3 , 5.5 , 5.6 , 5.7 )
- If patient develops symptoms consistent with peripheral neuropathy, evaluate patient and consider discontinuing teriflunomide tablets. (5.8 )
- Teriflunomide tablets may increase blood pressure. Measure blood pressure at treatment initiation and monitor blood pressure during treatment. (5.9 )
Hepatotoxicity
Clinically significant and potentially life-threatening liver injury, including acute liver failure requiring transplant, has been reported in patients treated with teriflunomide tablets in the postmarketing setting. Patients with pre-existing liver disease and patients taking other hepatotoxic drugs may be at increased risk for developing liver injury when taking teriflunomide tablets. Clinically significant liver injury can occur at any time during treatment with teriflunomide tablets.
Patients with pre-existing acute or chronic liver disease, or those with serum alanine aminotransferase (ALT) greater than two times the upper limit of normal (ULN) before initiating treatment, should not normally be treated with teriflunomide tablets. Teriflunomide tablets is contraindicated in patients with severe hepatic impairment [see Contraindications (4) ] .
In placebo-controlled trials in adult patients, ALT greater than three times the ULN occurred in 61/1045 (5.8%) and 62/1002 (6.2%) of patients receiving teriflunomide tablets 7 mg and 14 mg, respectively, and 38/997 (3.8%) of patients receiving placebo, during the treatment period. These elevations occurred mostly within the first year of treatment. Half of the cases returned to normal without drug discontinuation. In clinical trials, if ALT elevation was greater than three times the ULN on two consecutive tests, teriflunomide tablet was discontinued and patients underwent an accelerated elimination procedure [see Warnings and Precautions (5.3)]. Of the patients who underwent discontinuation and accelerated elimination in controlled trials, half returned to normal or near normal values within 2 months.
One patient in the controlled trials in adult patients developed ALT 32 times the ULN and jaundice 5 months after initiation of teriflunomide tablets 14 mg treatment. The patient was hospitalized for 5 weeks and recovered after plasmapheresis and cholestyramine accelerated elimination procedure.
Teriflunomide-induced liver injury in this patient could not be ruled out.
Obtain serum transaminase and bilirubin levels within 6 months before initiation of teriflunomide tablet therapy. Monitor ALT levels at least monthly for six months after starting teriflunomide tablets. Consider additional monitoring when teriflunomide tablet is given with other potentially hepatotoxic drugs.
Consider discontinuing teriflunomide tablets if serum transaminase increase (greater than three times the ULN) is confirmed. Monitor serum transaminase and bilirubin on teriflunomide tablet therapy, particularly in patients who develop symptoms suggestive of hepatic dysfunction, such as unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine. If liver injury is suspected to be teriflunomide-induced, discontinue teriflunomide tablets and start an accelerated elimination procedure [see Warnings and Precautions (5.3) ] and monitor liver tests weekly until normalized. If teriflunomide-induced liver injury is unlikely because some other probable cause has been found, resumption of teriflunomide tablet therapy may be considered.
Embryofetal Toxicity
Teriflunomide may cause fetal harm when administered to a pregnant woman. Teratogenicity and embryofetal lethality occurred in animal reproduction studies in multiple animal species at plasma teriflunomide exposures similar to or lower than that in humans at the maximum recommended human dose (MRHD) of 14 mg/day [see Use in Specific Populations (8.1) ] .
Teriflunomide tablet is contraindicated for use in pregnant women and in females of reproductive potential not using effective contraception [see Contraindications (4) ] . Exclude pregnancy before starting treatment with teriflunomide tablets in females of reproductive potential [see Dosage and Administration (2) ] . Advise females of reproductive potential to use effective contraception during teriflunomide tablets treatment and during an accelerated drug elimination procedure after teriflunomide tablets treatment [see Use in Specific Populations (8.3 )] . If a woman becomes pregnant while taking teriflunomide tablets, stop treatment with teriflunomide tablets, apprise the patient of the potential risk to a fetus, and perform an accelerated drug elimination procedure to achieve a plasma teriflunomide concentration of less than 0.02 mg/L [see Warnings and Precautions (5.3 )] .
Upon discontinuing teriflunomide tablets, it is recommended that all females of reproductive potential undergo an accelerated drug elimination procedure. Women receiving teriflunomide tablets treatment who wish to become pregnant must discontinue teriflunomide tablets and undergo an accelerated drug elimination procedure, which includes verification that plasma concentrations of teriflunomide are less than 0.02 mg/L (0.02 mcg/mL). Men wishing to father a child should also discontinue use of teriflunomide tablets and either undergo an accelerated elimination procedure or wait until verification that the plasma teriflunomide concentration is less than 0.02 mg/L (0.02 mcg/mL) [see Use in Specific Population (8.3 )] . Based on animal data, human plasma concentrations of teriflunomide of less than 0.02 mg/L (0.02 mcg/mL) are expected to have minimal embryofetal risk [see Contraindications (4 ), Warnings and Precautions (5.3 ), and Use in Specific Populations (8.1 )] .
Procedure for Accelerated Elimination of Teriflunomide
Teriflunomide is eliminated slowly from the plasma [see Clinical Pharmacology (12.3 )] . Without an accelerated elimination procedure, it takes on average 8 months to reach plasma concentrations less than 0.02 mg/L, although because of individual variations in drug clearance it may take as long as 2 years. An accelerated elimination procedure could be used at any time after discontinuation of teriflunomide tablets. Elimination can be accelerated by either of the following procedures:
- Administration of cholestyramine 8 g every 8 hours for 11 days. If cholestyramine 8 g three times a day is not well tolerated, cholestyramine 4 g three times a day can be used.
- Administration of 50 g oral activated charcoal powder every 12 hours for 11 days.
If either elimination procedure is poorly tolerated, treatment days do not need to be consecutive unless there is a need to lower teriflunomide plasma concentration rapidly.
At the end of 11 days, both regimens successfully accelerated teriflunomide elimination, leading to more than 98% decrease in teriflunomide plasma concentrations.
Use of the accelerated elimination procedure may potentially result in return of disease activity if the patient had been responding to teriflunomide tablet treatment.
Bone Marrow Effects/Immunosuppression Potential/Infections
Bone Marrow Effects A mean decrease compared to baseline in white blood cell (WBC) count of approximately 15% (mainly neutrophils and lymphocytes) and in platelet count of approximately 10% was observed in placebo-controlled trials in adult patients with 7 mg and 14 mg of teriflunomide tablets. The decrease in mean WBC count occurred during the first 6 weeks and WBC count remained low during treatment. In placebo-controlled studies in adult patient, neutrophil count < 1.5 x 10 9 /L was observed in 12% and 16% of patients receiving teriflunomide tablets 7 mg and 14 mg, respectively, compared with 7% of patients receiving placebo; lymphocyte count < 0.8 x 10 9 /L was observed in 10% and 12% of patients receiving teriflunomide tablets 7 mg and 14 mg, respectively, compared with 6% of patients receiving placebo. No cases of serious pancytopenia were reported in premarketing clinical trials of teriflunomide tablets but rare cases of pancytopenia and agranulocytosis have been reported in the postmarketing setting with leflunomide. A similar risk would be expected for teriflunomide tablets [see Clinical Pharmacology (12.3 )] . Cases of thrombocytopenia with teriflunomide tablets, including rare cases with platelet counts less than 50,000/mm 3 , have been reported in the postmarketing setting. Obtain a complete blood cell count (CBC) within 6 months before the initiation of treatment with teriflunomide tablets. Further monitoring should be based on signs and symptoms suggestive of bone marrow suppression.
Risk of Infection/Tuberculosis Screening Patients with active acute or chronic infections should not start treatment until the infection(s) is resolved. If a patient develops a serious infection consider suspending treatment with teriflunomide tablets and using an accelerated elimination procedure. Reassess the benefits and risks prior to resumption of therapy. Instruct patients receiving teriflunomide tablets to report symptoms of infections to a physician. Teriflunomide tablet is not recommended for patients with severe immunodeficiency, bone marrow disease, or severe, uncontrolled infections. Medications like teriflunomide tablets that have immunosuppression potential may cause patients to be more susceptible to infections, including opportunistic infections.
In placebo-controlled studies of teriflunomide tablets in adult patients, no overall increase in the risk of serious infections was observed with teriflunomide tablets 7 mg (2.2%) or 14 mg (2.7%) compared to placebo (2.2%).
However, one fatal case of klebsiella pneumonia sepsis occurred in a patient taking teriflunomide tablets 14 mg for 1.7 years. Fatal infections have been reported in the postmarketing setting in patients receiving leflunomide, especially Pneumocystis jirovecii pneumonia and aspergillosis. Most of the reports were confounded by concomitant immunosuppressant therapy and/or comorbid illness which, in addition to rheumatoid disease, may predispose patients to infection. In clinical studies with teriflunomide tablets, cytomegalovirus hepatitis reactivation has been observed.
In clinical studies with teriflunomide tablets in adult patients, cases of tuberculosis have been observed. Prior to initiating teriflunomide tablets, screen patients for latent tuberculosis infection with a tuberculin skin test or with a blood test for mycobacterium tuberculosis infection. Teriflunomide tablet has not been studied in patients with a positive tuberculosis screen, and the safety of teriflunomide tablets in individuals with latent tuberculosis infection is unknown. For patients testing positive in tuberculosis screening, treat by standard medical practice prior to therapy with teriflunomide tablets.
Vaccination No clinical data are available on the efficacy and safety of live vaccinations in patients taking teriflunomide tablets. Vaccination with live vaccines is not recommended. The long half-life of teriflunomide tablets should be considered when contemplating administration of a live vaccine after stopping teriflunomide tablets.
Malignancy The risk of malignancy, particularly lymphoproliferative disorders, is increased with the use of some immunosuppressive medications. There is a potential for immunosuppression with teriflunomide tablets. No apparent increase in the incidence of malignancies and lymphoproliferative disorders was reported in the teriflunomide tablets clinical trials, but larger and longer-term studies would be needed to determine whether there is an increased risk of malignancy or lymphoproliferative disorders with teriflunomide tablets.
Hypersensitivity Reactions
Teriflunomide tablets can cause anaphylaxis and severe allergic reactions [see Contraindications (4 )] . Signs and symptoms have included dyspnea, urticaria, and angioedema including lips, eyes, throat, and tongue.
Inform patients of the signs and symptoms of anaphylaxis and angioedema.
Serious Skin Reactions
Cases of serious skin reactions, sometimes fatal, including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS) [see Warnings and Precautions (5.7 )] , have been reported with teriflunomide tablets. Fatal outcomes were reported in one case of TEN and one case of DRESS.
Inform patients of the signs and symptoms that may signal a serious skin reaction. Instruct patients to discontinue teriflunomide tablets and seek immediate medical care should these signs and symptoms occur. Unless the reaction is clearly not drug related, discontinue teriflunomide tablets and begin an accelerated elimination procedure immediately [see Warnings and Precautions (5.3 )] . In such cases, patients should not be re-exposed to teriflunomide [see Contraindications (4 )] .
Drug Reaction with Eosinophilia and Systemic Symptoms
Drug reaction with eosinophilia and systemic symptoms (DRESS), also known as multiorgan hypersensitivity, has occurred with teriflunomide tablets. One fatal case of DRESS that occurred in close temporal association (34 days) with the initiation of teriflunomide tablets treatment has been reported in the postmarketing setting. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematologic abnormalities, myocarditis, or myositis, sometimes resembling an acute viral infection. Eosinophilia is often present. This disorder is variable in its expression, and other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity (e.g., fever, lymphadenopathy) may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately.
Discontinue teriflunomide tablets, unless an alternative etiology for the signs or symptoms is established, and begin an accelerated elimination procedure immediately [see Warnings and Precautions (5.3 )] . In such cases, patients should not be re-exposed to teriflunomide [see Contraindications (4 )] .
Peripheral Neuropathy
In placebo-controlled studies in adult patients, peripheral neuropathy, including both polyneuropathy and mononeuropathy (e.g., carpal tunnel syndrome), occurred more frequently in patients taking teriflunomide tablets than in patients taking placebo. The incidence of peripheral neuropathy confirmed by nerve conduction studies was 1.4% (13 patients) and 1.9% (17 patients) of patients receiving 7 mg and 14 mg of teriflunomide tablets, respectively, compared with 0.4% receiving placebo (4 patients). Treatment was discontinued in 0.7% (8 patients) with confirmed peripheral neuropathy (3 patients receiving teriflunomide tablets 7 mg and 5 patients receiving teriflunomide tablets 14 mg). Five of them recovered following treatment discontinuation. Not all cases of peripheral neuropathy resolved with continued treatment. Peripheral neuropathy also occurred in patients receiving leflunomide.
Age older than 60 years, concomitant neurotoxic medications, and diabetes may increase the risk for peripheral neuropathy . If a patient taking teriflunomide tablets develops symptoms consistent with peripheral neuropathy, such as bilateral numbness or tingling of hands or feet, consider discontinuing teriflunomide tablet therapy and performing an accelerated elimination procedure [see Warnings and Precautions (5.3 )] .
Increased Blood Pressure
In placebo-controlled studies in adult patients, the mean change from baseline to the end of study in systolic blood pressure was +2.3 mmHg and +2.7 mmHg for teriflunomide tablets 7 mg and 14 mg, respectively, and -0.6 mmHg for placebo. The change from baseline in diastolic blood pressure was +1.4 mmHg and +1.9 mmHg for teriflunomide tablets 7 mg and 14 mg, respectively, and -0.3 mmHg for placebo. Hypertension was an adverse reaction in 3.1% and 4.3% of patients treated with 7 mg or 14 mg of teriflunomide tablets compared with 1.8% for placebo. Check blood pressure before start of teriflunomide tablets treatment and periodically thereafter. Elevated blood pressure should be appropriately managed during treatment with teriflunomide tablets.
Respiratory Effects
Interstitial lung disease, including acute interstitial pneumonitis, has been reported with teriflunomide tablets in the postmarketing setting.
Interstitial lung disease and worsening of pre-existing interstitial lung disease have been reported during treatment with leflunomide. Interstitial lung disease may be fatal and may occur acutely at any time during therapy with a variable clinical presentation. New onset or worsening pulmonary symptoms, such as cough and dyspnea, with or without associated fever, may be a reason for discontinuation of therapy and for further investigation as appropriate. If discontinuation of the drug is necessary, consider initiation of an accelerated elimination procedure [see Warnings and Precautions (5.3 )] .
Pancreatitis in Pediatric Patients
Teriflunomide tablet is not approved for use in pediatric patients [ see Use in Specific Populations (8.4 ) ]. If pancreatitis is suspected, discontinue teriflunomide and start an accelerated elimination procedure [ see Warnings and Precautions (5.3 )] .
Pediatric information describing a clinical study in which efficacy was not demonstrated is approved for Sanofi-Aventis U.S. LLC’s Aubagio ® (teriflunomide) tablets. However, due to Sanofi-Aventis U.S. LLC’s marketing exclusivity rights, this drug product is not labeled with that information .
Concomitant Use with Immunosuppressive or Immunomodulating Therapies
Co-administration with antineoplastic or immunosuppressive therapies used for treatment of multiple sclerosis has not been evaluated. Safety studies in which teriflunomide tablet was concomitantly administered with other immune modulating therapies for up to one year (interferon beta, glatiramer acetate) did not reveal any specific safety concerns. The long term safety of these combinations in the treatment of multiple sclerosis has not been established.
In any situation in which the decision is made to switch from teriflunomide tablets to another agent with a known potential for hematologic suppression, it would be prudent to monitor for hematologic toxicity, because there will be overlap of systemic exposure to both compounds. Use of an accelerated elimination procedure may decrease this risk, but may also potentially result in return of disease activity if the patient had been responding to teriflunomide tablet treatment [see Warnings and Precautions (5.3 )] .
Risk of Allergic Reactions due to Tartrazine
The 7 mg strength tablets contain FD&C Yellow No. 5 (tartrazine) which may cause allergic-type reactions (including bronchial asthma) in certain susceptible persons. Although the overall incidence of FD&C Yellow No. 5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin hypersensitivity.
ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the prescribing information:
- Hepatotoxicity [see Contraindications (4 ) and Warnings and Precautions (5.1 )]
- Bone Marrow Effects/Immunosuppression Potential/Infections [see Warnings and Precautions (5.4 )]
- Hypersensitivity Reactions [see Contraindications (4 ) and Warnings and Precautions (5.5 )]
- Serious Skin Reactions [see Warnings and Precautions (5.6 )]
- Drug Reaction with Eosinophilia and Systemic Symptoms [see Warnings and Precautions (5.7 )]
- Peripheral Neuropathy [see Warnings and Precautions (5.8 )]
- Increased Blood Pressure [see Warnings and Precautions (5.9 )]
- Respiratory Effects [see Warnings and Precautions (5.10) ]
- Pancreatitis in Pediatric Patients [ see Warnings and Precautions (5.11 ) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
A total of 2047 patients receiving teriflunomide tablets (7 mg or 14 mg once daily) constituted the safety population in the pooled analysis of placebo-controlled studies in patients with relapsing forms of multiple sclerosis; of these, 71% were female. The average age was 37 years.
Table 1 lists adverse reactions in placebo-controlled trials with rates that were at least 2% for teriflunomide tablets patients and also at least 2% above the rate in placebo patients. The most common were headache, an increase in ALT, diarrhea, alopecia, and nausea. The adverse reaction most commonly associated with discontinuation was an increase in ALT (3.3%, 2.6%, and 2.3% of all patients in the teriflunomide tablets 7 mg, teriflunomide tablets 14 mg, and placebo treatment arms, respectively).
| Adverse Reaction | Teriflunomide Tablets 7 mg (N=1045) | Teriflunomide Tablets 14 mg (N=1002) | Placebo (N=997) |
| Headache | 18% | 16% | 15% |
| Increase in Alanine aminotransferase | 13% | 15% | 9% |
| Diarrhea | 13% | 14% | 8% |
| Alopecia | 10% | 13% | 5% |
| Nausea | 8% | 11% | 7% |
| Paresthesia | 8% | 9% | 7% |
| Arthralgia | 8% | 6% | 5% |
| Neutropenia | 4% | 6% | 2% |
| Hypertension | 3% | 4% | 2% |
Cardiovascular Deaths Four cardiovascular deaths, including three sudden deaths, and one myocardial infarction in a patient with a history of hyperlipidemia and hypertension were reported among approximately 2600 patients exposed to teriflunomide tablets in the premarketing database. These cardiovascular deaths occurred during uncontrolled extension studies, one to nine years after initiation of treatment. A relationship between teriflunomide tablets and cardiovascular death has not been established.
Acute Renal Failure In placebo-controlled studies, creatinine values increased more than 100% over baseline in 8/1045 (0.8%) patients in the 7 mg teriflunomide tablets group and 6/1002 (0.6%) patients in the 14 mg teriflunomide tablets group versus 4/997 (0.4%) patients in the placebo group. These elevations were transient. Some elevations were accompanied by hyperkalemia. Teriflunomide tablets may cause acute uric acid nephropathy with transient acute renal failure because teriflunomide tablets increases renal uric acid clearance.
Hypophosphatemia In clinical trials, 18% of teriflunomide tablet-treated patients had hypophosphatemia with serum phosphorus levels of at least 0.6 mmol/L, compared to 7% of placebo-treated patients; 4% of teriflunomide tablet-treated patients had hypophosphatemia with serum phosphorus levels at least 0.3 mmol/L but less than 0.6 mmol/L, compared to 0.8% of placebo-treated patients. No patient in any treatment group had a serum phosphorus below 0.3 mmol/L.
Postmarketing Experience
The following adverse reactions have been identified during postapproval use of teriflunomide tablets. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Blood and Lymphatic System Disorders : Thrombocytopenia [see Warnings and Precautions (5.4 )]
- Gastrointestinal Disorders : Pancreatitis, colitis
- Hepatobiliary Disorders : Drug-induced liver injury (DILI) [see Warnings and Precautions (5.1 )]
- Immune System Disorders : Hypersensitivity reactions, some of which were severe, such as anaphylaxis and angioedema [see Warnings and Precautions (5.5 )]
- Respiratory, Thoracic, and Mediastinal Disorders : Interstitial lung disease [see Warnings and Precautions (5.10 )]
- Skin and Subcutaneous Tissue Disorders : Severe skin reactions, including toxic epidermal necrolysis and Stevens-Johnson syndrome [see Warnings and Precautions (5.6 )] ; drug reaction with eosinophilia and systemic symptoms (DRESS) [see Warnings and Precautions (5.7 )] ; psoriasis or worsening of psoriasis (including pustular psoriasis and nail psoriasis); nail disorders
DRUG INTERACTIONS
Effect of Teriflunomide Tablets on CYP2C8 Substrates Teriflunomide is an inhibitor of CYP2C8 in vivo . In patients taking teriflunomide tablets, exposure of drugs metabolized by CYP2C8 (e.g., paclitaxel, pioglitazone, repaglinide, rosiglitazone) may be increased. Monitor these patients and adjust the dose of the concomitant drug(s) metabolized by CYP2C8 as required [see Clinical Pharmacology (12.3 )] .
Effect of Teriflunomide Tablets on Warfarin Coadministration of teriflunomide tablets with warfarin requires close monitoring of the international normalized ratio (INR) because teriflunomide tablets may decrease peak INR by approximately 25%.
Effect of Teriflunomide Tablets on Oral Contraceptives Teriflunomide tablets may increase the systemic exposures of ethinylestradiol and levonorgestrel. Consideration should be given to the type or dose of contraceptives used in combination with teriflunomide tablets [see Clinical Pharmacology (12.3 )] .
Effect of Teriflunomide Tablets on CYP1A2 Substrates Teriflunomide may be a weak inducer of CYP1A2 in vivo . In patients taking teriflunomide tablets, exposure of drugs metabolized by CYP1A2 (e.g., alosetron, duloxetine, theophylline, tizanidine) may be reduced. Monitor these patients and adjust the dose of the concomitant drug(s) metabolized by CYP1A2 as required [see Clinical Pharmacology (12.3 )] .
Effect of Teriflunomide Tablets on Organic Anion Transporter 3 (OAT3) Substrates Teriflunomide inhibits the activity of OAT3 in vivo . In patients taking teriflunomide tablets, exposure of drugs which are OAT3 substrates (e.g., cefaclor, cimetidine, ciprofloxacin, penicillin G, ketoprofen, furosemide, methotrexate, zidovudine) may be increased. Monitor these patients and adjust the dose of the concomitant drug(s) which are OAT3 substrates as required [see Clinical Pharmacology (12.3 )] .
Effect of Teriflunomide Tablets on BCRP and Organic Anion Transporting Polypeptide B1 and B3 (OATP1B1/1B3) Substrates Teriflunomide inhibits the activity of BCRP and OATP1B1/1B3 in vivo . For a patient taking teriflunomide tablets, the dose of rosuvastatin should not exceed 10 mg once daily. For other substrates of BCRP (e.g., mitoxantrone) and drugs in the OATP family (e.g., methotrexate, rifampin), especially HMG-Co reductase inhibitors (e.g., atorvastatin, nateglinide, pravastatin, repaglinide, and simvastatin), consider reducing the dose of these drugs and monitor patients closely for signs and symptoms of increased exposures to the drugs while patients are taking teriflunomide tablets [see Clinical Pharmacology (12.3 )] .
DESCRIPTION
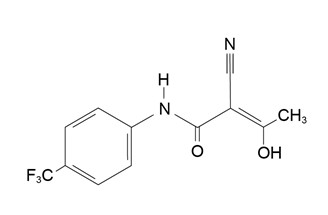
Teriflunomide is an oral de novo pyrimidine synthesis inhibitor of the DHO-DH enzyme, with the chemical name (Z)-2-Cyano-3-hydroxy-but-2-enoic acid-(4 trifluoromethylphenyl)-amide. Its molecular weight is 270.21, and the empirical formula is C 12 H 9 F 3 N 2 O 2 with the following chemical structure:
Teriflunomide is a white to almost white powder that is sparingly soluble in acetone, slightly soluble in polyethylene glycol and ethanol, very slightly soluble in isopropanol and practically insoluble in water.
Teriflunomide is formulated as film-coated tablets for oral administration. Teriflunomide tablets contain 7 mg and 14 mg of teriflunomide and the following inactive ingredients: lactose monohydrate, hydroxypropylcellulose, microcrystalline cellulose, colloidal silicon dioxide, and magnesium stearate. The film coating for the 7mg and 14mg tablets is made of hypromellose, hydroxypropylcellulose, cottonseed oil, talcum, titanium dioxide, and FD&C Blue no. 2 Al-Lake. In addition to these, the 7mg tablet film coating includes FD&C Yellow no. 5 (tartrazine). The 14mg tablet includes FD&C Red no. 40.
CLINICAL PHARMACOLOGY
Mechanism of Action
Teriflunomide, an immunomodulatory agent with anti-inflammatory properties, inhibits dihydroorotate dehydrogenase, a mitochondrial enzyme involved in de novo pyrimidine synthesis. The exact mechanism by which teriflunomide exerts its therapeutic effect in multiple sclerosis is unknown but may involve a reduction in the number of activated lymphocytes in CNS.
Pharmacodynamics
Potential to Prolong the QT Interval In a placebo controlled thorough QT study performed in healthy adult subjects, there was no evidence that teriflunomide caused QT interval prolongation of clinical significance (i.e., the upper bound of the 90% confidence interval for the largest placebo-adjusted, baseline-corrected QTc was below 10 ms).
Pharmacokinetics
Teriflunomide is the principal active metabolite of leflunomide and is responsible for leflunomide’s activity in vivo . At recommended doses, teriflunomide and leflunomide result in a similar range of plasma concentrations of teriflunomide.
Based on a population analysis of teriflunomide in healthy adult volunteers and adult MS patients, median t 1/2 was approximately 18 and 19 days after repeated doses of 7 mg and 14 mg respectively. It takes approximately 3 months respectively to reach steady-state concentrations. The estimated AUC accumulation ratio is approximately 30 after repeated doses of 7 or 14 mg.
Absorption Median time to reach maximum plasma concentrations is between 1 to 4 hours post dose following oral administration of teriflunomide. Food does not have a clinically relevant effect on teriflunomide pharmacokinetics.
Distribution Teriflunomide is extensively bound to plasma protein (>99%) and is mainly distributed in plasma. The volume of distribution is 11 L after a single intravenous (IV) administration.
Metabolism Teriflunomide is the major circulating moiety detected in plasma. The primary biotransformation pathway to minor metabolites of teriflunomide is hydrolysis, with oxidation being a minor pathway. Secondary pathways involve oxidation, N-acetylation and sulfate conjugation.
Elimination Teriflunomide is eliminated mainly through direct biliary excretion of unchanged drug as well as renal excretion of metabolites. Over 21 days, 60.1% of the administered dose is excreted via feces (37.5%) and urine (22.6%). After an accelerated elimination procedure with cholestyramine, an additional 23.1% was recovered (mostly in feces). After a single IV administration, the total body clearance of teriflunomide is 30.5 mL/h.
Drug Interaction Studies Teriflunomide is not metabolized by Cytochrome P450 or flavin monoamine oxidase enzymes. The potential effect of teriflunomide tablets on other drugs
CYP2C8 substrates
There was an increase in mean repaglinide C max and AUC (1.7- and 2.4-fold, respectively) following repeated doses of teriflunomide and a single dose of 0.25 mg repaglinide, suggesting that teriflunomide is an inhibitor of CYP2C8 in vivo . The magnitude of interaction could be higher at the recommended repaglinide dose [see Drug Interactions (7) ] .
CYP1A2 substrates
Repeated doses of teriflunomide decreased mean C max and AUC of caffeine by 18% and 55%, respectively, suggesting that teriflunomide may be a weak inducer of CYP1A2 in vivo [see Drug Interactions (7 )].
OAT3 substrates
There was an increase in mean cefaclor C max and AUC (1.43- and 1.54-fold, respectively), following repeated doses of teriflunomide, suggesting that teriflunomide is an inhibitor of organic anion transporter 3 (OAT3) in vivo [see Drug Interactions (7) ] .
BCRP and OATP1B1/1B3 substrates
There was an increase in mean rosuvastatin C max and AUC (2.65- and 2.51-fold, respectively) following repeated doses of teriflunomide, suggesting that teriflunomide is an inhibitor of BCRP transporter and organic anion transporting polypeptide 1B1 and 1B3 (OATP1B1/1B3) [see Drug Interactions (7) ] .
Oral contraceptives
There was an increase in mean ethinylestradiol C max and AUC 0-24 (1.58- and 1.54-fold, respectively) and levonorgestrel C max and AUC 0-24 (1.33- and 1.41-fold, respectively) following repeated doses of teriflunomide [see Drug Interactions (7) ] .
Teriflunomide did not affect the pharmacokinetics of bupropion (a CYP2B6 substrate), midazolam (a CYP3A4 substrate), S-warfarin (a CYP2C9 substrate), omeprazole (a CYP2C19 substrate), and metoprolol (a CYP2D6 substrate).
The potential effect of other drugs on teriflunomide tablets
Potent CYP and transporter inducers: Rifampin did not affect the pharmacokinetics of teriflunomide.
Specific Populations
Hepatic impairment
Mild and moderate hepatic impairment had no impact on the pharmacokinetics of teriflunomide. The pharmacokinetics of teriflunomide in severe hepatic impairment has not been evaluated [see Contraindications (4 ), Warnings and Precautions (5.1 ), and Use in Specific Populations (8.6 )] .
Renal impairment
Severe renal impairment had no impact on the pharmacokinetics of teriflunomide [see Use in Specific Populations (8.7 )] .
Gender
In a population analysis, the clearance rate for teriflunomide is 23% less in females than in males.
Race
Effect of race on the pharmacokinetics of teriflunomide cannot be adequately assessed due to a low number of non-white patients in the clinical trials.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No evidence of carcinogenicity was observed in lifetime carcinogenicity bioassays in mouse and rat. In mouse, teriflunomide was administered orally at doses up to 12 mg/kg/day for up to 95 to 104 weeks; plasma teriflunomide exposures (AUC) at the highest dose tested are approximately 3 times that in humans at the maximum recommended human dose (MRHD, 14 mg/day). In rat, teriflunomide was administered orally at doses up to 4 mg/kg/day for up to 97 to 104 weeks; plasma teriflunomide AUCs at the highest doses tested are less than that in humans at the MRHD.
Mutagenesis
Teriflunomide was negative in the in vitro bacterial reverse mutation (Ames) assay, the in vitro HPRT assay, and in in vivo micronucleus and chromosomal aberration assays. Teriflunomide was positive in an in vitro chromosomal aberration assay in human lymphocytes, with and without metabolic activation. Addition of uridine (to supplement the pyrimidine pool) reduced the magnitude of the clastogenic effect; however, teriflunomide was positive in the in vitro chromosomal aberration assay, even in the presence of uridine.
4-Trifluoromethylaniline (4-TFMA), a minor metabolite of teriflunomide, was positive in the in vitro bacterial reverse mutation (Ames) assay, the in vitro HPRT assay, and the in vitro chromosomal aberration assay in mammalian cells. 4-TFMA was negative in in vivo micronucleus and chromosomal aberration assays.
Impairment of Fertility
Oral administration of teriflunomide (0, 1, 3, 10 mg/kg/day) to male rats prior to and during mating (to untreated females) resulted in no adverse effects on fertility; however, reduced epididymal sperm count was observed at the mid and high doses tested. The no-effect dose for reproductive toxicity in male rats (1 mg/kg) is less than the MRHD on a mg/m 2 basis.
Oral administration of teriflunomide (0, 0.84, 2.6, 8.6 mg/kg/day) to female rats, prior to and during mating (to untreated males) and continuing to gestation day 6, resulted in embryolethality, reduced fetal body weight, and/or malformations at all doses tested. Due to marked embryolethality at the highest dose tested, no fetuses were available for evaluation. The lowest dose tested is less than the MRHD on a mg/m 2 basis.
CLINICAL STUDIES
Four randomized, controlled, double-blind clinical trials established the efficacy of teriflunomide tablets in patients with relapsing forms of multiple sclerosis.
Study 1 was a double-blind, placebo-controlled clinical trial that evaluated once daily doses of teriflunomide tablets 7 mg and teriflunomide tablets 14 mg for up to 26 months in patients with relapsing forms of multiple sclerosis. Patients were required to have a diagnosis of multiple sclerosis exhibiting a relapsing clinical course, with or without progression, and to have experienced at least one relapse over the year preceding the trial or at least two relapses over the two years preceding the trial. Patients were required not to have received interferon-beta for at least four months, or any other multiple sclerosis medication for at least six months before entering the study, nor were these medications permitted during the study. Neurological evaluations were to be performed at screening, every 12 weeks until week 108, and after suspected relapses. MRI was to be performed at screening, and at Week 24, 48, 72, and 108. The primary endpoint was the annualized relapse rate (ARR).
In Study 1, 1088 patients were randomized to receive teriflunomide tablets 7 mg (n=366), teriflunomide tablets 14 mg (n=359), or placebo (n=363). At entry, patients had an Expanded Disability Status Scale (EDSS) score ≤5.5. Patients had a mean age of 38 years, mean disease duration of 5 years, and mean EDSS at baseline of 2.7. A total of 91% of patients had relapsing remitting multiple sclerosis, and 9% had a progressive form of multiple sclerosis with relapses. The mean duration of treatment was 635, 627, and 631 days for teriflunomide tablets 7 mg, teriflunomide tablets 14 mg, and placebo, respectively. The percentage of patients who completed the study treatment period was 75%, 73%, and 71% for teriflunomide tablets 7 mg, teriflunomide tablets 14 mg, and placebo, respectively.
There was a statistically significant reduction in ARR for patients who received teriflunomide tablets 7 mg or teriflunomide tablets 14 mg, compared to patients who received placebo (see Table 2 ). There was a consistent reduction of the ARR noted in subgroups defined by sex, age group, prior multiple sclerosis therapy, and baseline disease activity.
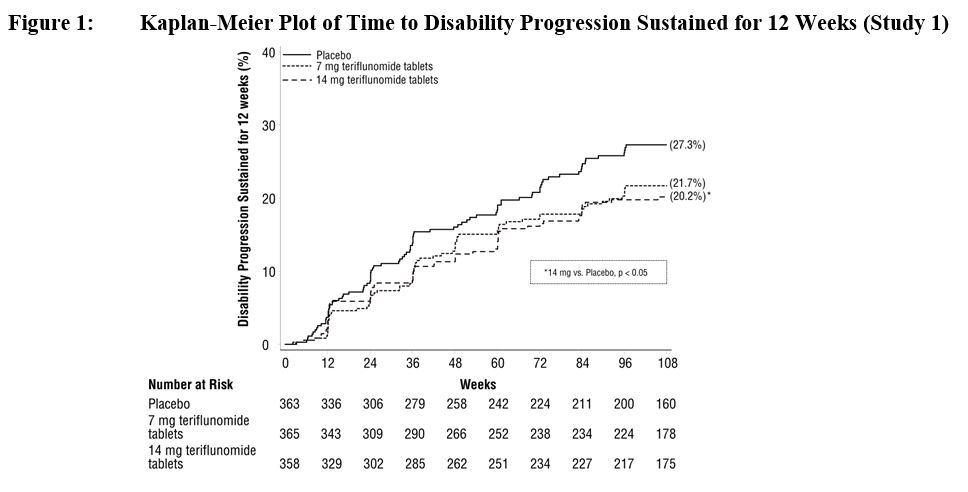
There was a statistically significant reduction in the relative risk of disability progression at week 108 sustained for 12 weeks (as measured by at least a 1-point increase from baseline EDSS ≤ 5.5 or a 0.5 point increase for those with a baseline EDSS > 5.5) in the teriflunomide tablets 14 mg group compared to placebo (see Table 2 and Figure 1 ).
The effect of teriflunomide tablets on several magnetic resonance imaging (MRI) variables, including the total lesion volume of T2 and hypointense T1 lesions, was assessed in Study 1. The change in total lesion volume from baseline was significantly lower in the teriflunomide tablets 7 mg and teriflunomide tablets 14 mg groups than in the placebo group. Patients in both teriflunomide tablets groups had significantly fewer gadolinium-enhancing lesions per T1-weighted scan than those in the placebo group (see Table 2 ).
| Teriflunomide Tablets 7 mg N=365 | Teriflunomide Tablets 14 mg N=358 | Placebo N=363 | |
| 1 Total lesion volume: sum of T2 and hypointense T1 lesion volume in mL 2 p-values based on cubic root transformed data for total lesion volume | |||
| Clinical Endpoints | |||
| Annualized relapse rate | 0.370 (p=0.0002) | 0.369 (p=0.0005) | 0.539 |
| Relative risk reduction | 31% | 31% | – |
| Percent of patients remaining relapse-free at week 108 | 53.7% | 56.5% | 45.6% |
| Percent disability progression at week 108 | 21.7% (p=0.084) | 20.2% (p=0.028) | 27.3% |
| Hazard ratio | 0.76 | 0.70 | – |
| MRI Endpoints | |||
| Median change from baseline in Total lesion volume 1 (mL) at week 108 | 0.755 (p=0.0317) 2 | 0.345 (p=0.0003) 2 | 1.127 |
| Mean number of Gd-enhancing T1-lesions per scan | 0.570 (p<0.0001) | 0.261 (p<0.0001) | 1.331 |
Study 2 was a double-blind, placebo-controlled clinical trial that evaluated once daily doses of teriflunomide tablets 7 mg and teriflunomide tablets 14 mg for up to 40 months in patients with relapsing forms of multiple sclerosis. Patients were required to have a diagnosis of multiple sclerosis exhibiting a relapsing clinical course and to have experienced at least one relapse over the year preceding the trial, or at least two relapses over the two years preceding the trial. Patients were required not to have received any multiple sclerosis medication for at least three months before entering the trial, nor were these medications permitted during the trial. Neurological evaluations were to be performed at screening, every 12 weeks until completion, and after every suspected relapse. The primary end point was the ARR.
A total of 1165 patients received teriflunomide tablets 7 mg (n=407), teriflunomide tablets 14 mg (n=370), or placebo (n=388). Patients had a mean age of 38 years, a mean disease duration of 5 years, and a mean EDSS at baseline of 2.7. A total of 98% of patients had relapsing remitting multiple sclerosis, and 2% had a progressive form of multiple sclerosis with relapses. The mean duration of treatment was 552, 567, and 571 days for teriflunomide tablets 7 mg, teriflunomide tablets 14 mg, and placebo, respectively. The percentage of patients who completed the study treatment period was 67%, 66%, and 68% for teriflunomide tablets 7 mg, teriflunomide tablets 14 mg, and placebo, respectively.
There was a statistically significant reduction in the ARR for patients who received teriflunomide tablets 7 mg or teriflunomide tablets 14 mg compared to patients who received placebo (see Table 3 ). There was a consistent reduction of the ARR noted in subgroups defined by sex, age group, prior multiple sclerosis therapy, and baseline disease activity.
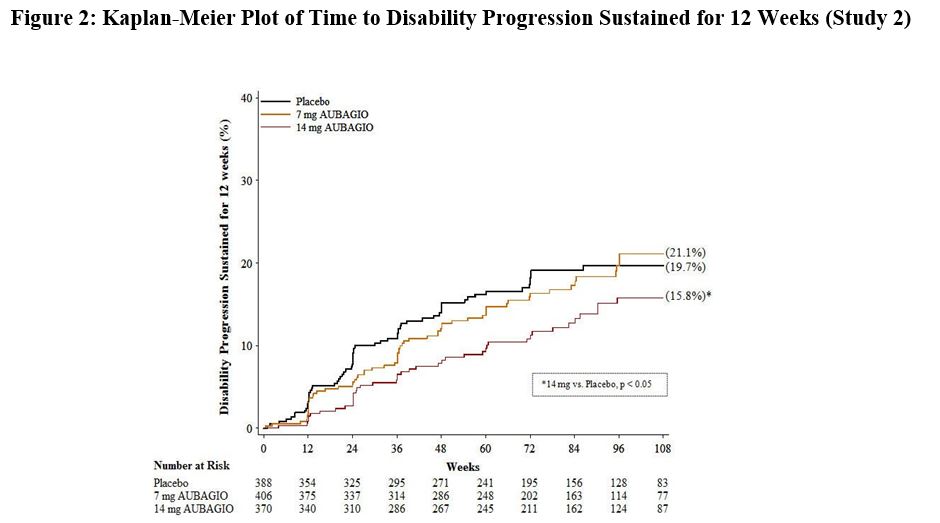
There was a statistically significant reduction in the relative risk of disability progression at week 108 sustained for 12 weeks (as measured by at least a 1-point increase from baseline EDSS ≤ 5.5 or a 0.5 point increase for those with a baseline EDSS > 5.5) in the teriflunomide tablets 14 mg group compared to placebo (see Table 3 and Figure 2 ).
| Teriflunomide Tablets 7 mg N=407 | Teriflunomide Tablets 14 mg N=370 | Placebo N=388 | |
| Clinical Endpoints | |||
| Annualized relapse rate | 0.389 (p=0.0183) | 0.319 (p=0.0001) | 0.501 |
| Relative risk reduction | 22% | 36% | – |
| Percent of patients remaining relapse-free at week 108 | 58.2% | 57.1% | 46.8% |
| Percent disability progression at week 108 | 21.1% (p=0.762) | 15.8% (p=0.044) | 19.7% |
| Hazard ratio | 0.96 | 0.69 | – |
Study 3 was a double-blind, placebo-controlled clinical trial that evaluated once daily doses of teriflunomide tablets 7 mg and teriflunomide tablets 14 mg for up to 108 weeks in patients with relapsing multiple sclerosis. Patients were required to have had a first clinical event consistent with acute demyelination occurring within 90 days of randomization with 2 or more T2 lesions at least 3 mm in diameter that were characteristic of multiple sclerosis. A total of 614 patients received teriflunomide tablets 7 mg (n=203), teriflunomide tablets 14 mg (n=214), or placebo (n=197). Patients had a mean age of 32 years, EDSS at baseline of 1.7, and mean disease duration of two months. The proportion of patients free of relapse was greater in the teriflunomide tablets 7 mg (70.5%, p <0.05) and teriflunomide tablets 14 mg (72.2%, p <0.05) groups than in the placebo group (61.7%).
The effect of teriflunomide tablets on MRI activity was also demonstrated in Study 4, a randomized, double-blind, placebo-controlled clinical trial of multiple sclerosis patients with relapse. In Study 4, MRI was to be performed at baseline, 6 weeks, 12 weeks, 18 weeks, 24 weeks, 30 weeks, and 36 weeks after treatment initiation. A total of 179 patients were randomized to teriflunomide tablets 7 mg (n=61), teriflunomide tablets 14 mg (n=57), or placebo (n=61). Baseline demographics were consistent across treatment groups. The primary endpoint was the average number of unique active lesions/MRI scan during treatment. The mean number of unique active lesions per brain MRI scan during the 36-week treatment period was lower in patients treated with teriflunomide tablets 7 mg (1.06) and teriflunomide tablets 14 mg (0.98) as compared to placebo (2.69), the difference being statistically significant for both (p=0.0234 and p=0.0052, respectively).
HOW SUPPLIED/STORAGE AND HANDLING
Teriflunomide tablets are available as 7 mg and 14 mg tablets.
The 14 mg tablet is a blue (pale blue to pastel blue) colored, round, biconvex, film-coated tablet debossed with the dose strength “14” on one side. Each tablet contains 14 mg of teriflunomide.
The 7 mg tablet is a green (light greenish-blue grey to pale greenish-blue) colored, round, biconvex, film-coated tablet debossed with the dose strength “7” on one side. Each tablet contains 7 mg of teriflunomide.
Teriflunomide 14 mg tablets are supplied as: NDC 70512-0851-28 Carton of 28 tablets containing 1 wallet composed of 2 folded blister cards of 14 tablets per blister card
Teriflunomide 7 mg tablets are supplied as: NDC 70512-0850-28 Carton of 28 tablets containing 1 wallet composed of 2 folded blister cards of 14 tablets per blister card
Store at 68°F to 77°F (20°C to 25°C) [see USP Controlled Room Temperature] with excursions permitted between 59°F and 86°F (15°C and 30°C).
Mechanism of Action
Teriflunomide, an immunomodulatory agent with anti-inflammatory properties, inhibits dihydroorotate dehydrogenase, a mitochondrial enzyme involved in de novo pyrimidine synthesis. The exact mechanism by which teriflunomide exerts its therapeutic effect in multiple sclerosis is unknown but may involve a reduction in the number of activated lymphocytes in CNS.