Get your patient on Tolcapone - Tolcapone tablet (Tolcapone)
Tolcapone - Tolcapone tablet prescribing information
WARNING
Because of the risk of potentially fatal, acute fulminant liver failure, tolcapone tablets should ordinarily be used in patients with Parkinson's disease on l-dopa/carbidopa who are experiencing symptom fluctuations and are not responding satisfactorily to or are not appropriate candidates for other adjunctive therapies (see INDICATIONS AND USAGE and DOSAGE AND ADMINISTRATION sections).
Because of the risk of liver injury and because tolcapone tablets, when it is effective, provides an observable symptomatic benefit, the patient who fails to show substantial clinical benefit within 3 weeks of initiation of treatment, should be withdrawn from tolcapone tablets.
Tolcapone tablets therapy should not be initiated if the patient exhibits clinical evidence of liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. Patients with severe dyskinesia or dystonia should be treated with caution (see PRECAUTIONS: Rhabdomyolysis ).
PATIENTS WHO DEVELOP EVIDENCE OF HEPATOCELLULAR INJURY WHILE ON TOLCAPONE TABLETS AND ARE WITHDRAWN FROM THE DRUG FOR ANY REASON MAY BE AT INCREASED RISK FOR LIVER INJURY IF TOLCAPONE TABLETS IS REINTRODUCED. ACCORDINGLY, SUCH PATIENTS SHOULD NOT ORDINARILY BE CONSIDERED FOR RETREATMENT.
Cases of severe hepatocellular injury, including fulminant liver failure resulting in death, have been reported in postmarketing use. As of May 2005, 3 cases of fatal fulminant hepatic failure have been reported from more than 40,000 patient years of worldwide use. This incidence may be 10-to100-fold higher than the background incidence in the general population. Underreporting of cases may lead to significant underestimation of the increased risk associated with the use of tolcapone tablets. All 3 cases were reported within the first six months of initiation of treatment with tolcapone tablets. Analysis of the laboratory monitoring data in over 3,400 tolcapone tablets-treated patients participating in clinical trials indicated that increases in SGPT/ALT or SGOT/AST, when present,generally occurred within the first 6 months of treatment with tolcapone tablets.
A prescriber who elects to use tolcapone tablets in face of the increased risk of liver injury is strongly advised to monitor patients for evidence of emergent liver injury. Patients should be advised of the need for self-monitoring for both the classical signs of liver disease (e.g., clay colored stools, jaundice) and the nonspecific ones (eg, fatigue, loss of appetite, lethargy).
Although a program of periodic laboratory monitoring for evidence of hepatocellular injury is recommended, it is not clear that periodic monitoring of liver enzymes will prevent the occurrence of fulminant liver failure. However, it is generally believed that early detection of drug-induced hepatic injury along with immediate withdrawal of the suspect drug enhances the likelihood for recovery. Accordingly, the following liver monitoring program is recommended.
Before starting treatment with tolcapone tablets, the physician should conduct appropriate tests to exclude the presence of liver disease. In patients determined to be appropriate candidates for treatment with tolcapone tablets, serum glutamic-pyruvic transaminase (SGPT/ALT) and serum glutamic-oxaloacetic transaminase (SGOT/AST) levels should be determined at baseline and periodically (i.e. every 2 to 4 weeks) for the first 6 months of therapy. After the first six months, periodic monitoring is recommended at intervals deemed clinically relevant. Although more frequent monitoring increases the chances of early detection, the precise schedule for monitoring is a matter of clinical judgment. If the dose is increased to 200 mg tid (see DOSAGE AND ADMINISTRATION section), liver enzyme monitoring should take place before increasing the dose and then be conducted every 2 to 4 weeks for the following 6 months of therapy. After six months, periodic monitoring is recommended at intervals deemed clinically relevant.
Tolcapone tablets should be discontinued if SGPT/ALT or SGOT/AST levels exceed 2 times the upper limit of normal or if clinical signs and symptoms suggest the onset of hepatic dysfunction (persistent nausea, fatigue, lethargy, anorexia, jaundice, dark urine, pruritus, and right upper quadrant tenderness).
INDICATIONS AND USAGE
Tolcapone tablets, USP is indicated as an adjunct to levodopa and carbidopa for the treatment of the signs and symptoms of idiopathic Parkinson's disease. Because of the risk of potentially fatal, acute fulminant liver failure, tolcapone tablets, USP should ordinarily be used in patients with Parkinson's disease on l-dopa/carbidopa who are experiencing symptom fluctuations and are not responding satisfactorily to or are not appropriate candidates for other adjunctive therapies. Because of the risk of liver injury and because tolcapone tablets, USP, when it is effective, provides an observable symptomatic benefit, the patient who fails to show substantial clinical benefit within 3 weeks of initiation of treatment, should be withdrawn from tolcapone tablets, USP.
The effectiveness of tolcapone tablets, USP was demonstrated in randomized controlled trials in patients receiving concomitant levodopa therapy with carbidopa or another aromatic amino acid decarboxylase inhibitor who experienced end of dose wearing-off phenomena as well as in patients who did not experience such phenomena (see CLINICAL PHARMACOLOGY : Clinical Studies ).
DOSAGE AND ADMINISTRATION
Because of the risk of potentially fatal, acute fulminant liver failure, tolcapone tablets should ordinarily be used in patients with Parkinson's disease on l-dopa/carbidopa who are experiencing symptom fluctuations and are not responding satisfactorily to or are not appropriate candidates for other adjunctive therapies (see INDICATIONS AND USAGE and DOSAGE AND ADMINISTRATION sections).
BECAUSE OF THE RISK OF LIVER INJURY AND BECAUSE TOLCAPONE TABLETS WHEN IT IS EFFECTIVE PROVIDES AN OBSERVABLE SYMPTOMATIC BENEFIT, THE PATIENT WHO FAILS TO SHOW SUBSTANTIAL CLINICAL BENEFIT WITHIN 3 WEEKS OF INITIATION OF TREATMENT, SHOULD BE WITHDRAWN FROM TOLCAPONE TABLETS
Tolcapone tablets therapy should not be initiated if the patient exhibits clinical evidence of liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. Patients with severe dyskinesia or dystonia should be treated with caution (see PRECAUTIONS : Rhabdomyolysis).
Patients who develop evidence of hepatocellular injury while on tolcapone tablets and are withdrawn from the drug for any reason may be at increased risk for liver injury if tolcapone tablets is reintroduced. These patients should not ordinarily be considered for retreatment with tolcapone tablets.
Only prescribe tolcapone tablets for patients taking concomitant carbidopa levodopa therapy. The initial dose of tolcapone tablets is always 100 mg three times per day. The recommended daily dose of tolcapone tablets is also 100 mg tid. In clinical trials, elevations in ALT occurred more frequently at the dose of 200 mg tid. While it is unknown whether the risk of acute fulminant liver failure is increased at the 200-mg dose, it would be prudent to use 200 mg only if the anticipated incremental clinical benefit is justified (see BOXED WARNING , WARNINGS , PRECAUTIONS : Laboratory Tests ). If a patient fails to show the expected incremental benefit on the 200-mg dose after a total of 3 weeks of treatment (regardless of dose), tolcapone tablets should be discontinued.
In clinical trials, the first dose of the day of tolcapone tablets was always taken together with the first dose of the day of levodopa/carbidopa, and the subsequent doses of tolcapone tablets were given approximately 6 and 12 hours later.
In clinical trials, the majority of patients required a decrease in their daily levodopa dose if their daily dose of levodopa was >600 mg or if patients had moderate or severe dyskinesias before beginning treatment.
To optimize an individual patient's response, reductions in daily levodopa dose may be necessary. In clinical trials, the average reduction in daily levodopa dose was about 30% in those patients requiring a levodopa dose reduction. (Greater than 70% of patients with levodopa doses above 600 mg daily required such a reduction.)
Tolcapone tablets can be combined with both the immediate and sustained release formulations of levodopa/carbidopa.
Tolcapone tablets may be taken with or without food (see CLINICAL PHARMACOLOGY ).
Patients With Impaired Hepatic Function: Tolcapone tablets therapy should not be initiated in any patient with liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal. (See BOXED WARNING , WARNINGS , and CLINICAL PHARMACOLOGY ).
Patients With Impaired Renal Function: No dose adjustment of tolcapone tablets is recommended for patients with mild to moderate renal impairment. However, patients with severe renal impairment should be treated with caution. The safety of tolcapone has not been examined in subjects who had creatinine clearance less than 25 mL/min (see CLINICAL PHARMACOLOGY ).
Withdrawing Patients From Tolcapone Tablets: As with any dopaminergic drug, withdrawal or abrupt reduction in the tolcapone tablets dose may lead to emergence of signs and symptoms of Parkinson's disease or Hyperpyrexia and Confusion, a syndrome complex resembling the neuroleptic malignant syndrome (see PRECAUTIONS : Events Reported With Dopaminergic Therapy ). If a decision is made to discontinue treatment with tolcapone tablets, then it is recommended to closely monitor the patient and adjust other dopaminergic treatments as needed. This syndrome should be considered in the differential diagnosis for any patient who develops a high fever or severe rigidity. Tapering tolcapone tablets has not been systematically evaluated. As the duration of COMT inhibition with tolcapone tablets is generally 5 to 6 hours on average, decreasing the frequency of dosage to twice or once a day may not in itself prevent withdrawal effects.
CONTRAINDICATIONS
Tolcapone tablets are contraindicated in patients with liver disease, in patients who were withdrawn from tolcapone tablets because of evidence of tolcapone tablets-induced hepatocellular injury or who have demonstrated hypersensitivity to the drug or its ingredients.
Tolcapone tablets is also contraindicated in patients with a history of nontraumatic rhabdomyolysis or hyperpyrexia and confusion possibly related to medication (see PRECAUTIONS : Events Reported With Dopaminergic Therapy ).
ADVERSE REACTIONS
Cases of severe hepatocellular injury, including fulminant liver failure resulting in death, have been reported in postmarketing use. As of May 2005, 3 cases of fatal fulminant hepatic failure have been reported from more than 40,000 patient years of worldwide use. This incidence may be 10- to 100- fold higher than the background incidence in the general population. All 3 cases were reported within the first six months of initiation of treatment with tolcapone tablets. Analysis of the laboratory monitoring data in over 3,400 tolcapone tablets-treated patients participating in clinical trials indicated that increases in SGPT/ALT or SGOT/AST, when present, generally occurred within the first 6 months of treatment with tolcapone tablets.
The imprecision of the estimated increase is due to uncertainties about the base rate and the actual number of cases occurring in association with tolcapone tablets. The incidence of idiopathic potentially fatal fulminant hepatic failure (i.e., not due to viral hepatitis or alcohol) is low. One estimate, based upon transplant registry data, is approximately 3/1,000,000 patients per year in the United States. Whether this estimate is an appropriate basis for estimating the increased risk of liver failure among tolcapone tablets users is uncertain. Tolcapone tablets users, for example, differ in age and general health status from candidates for liver transplantation. Similarly, underreporting of cases may lead to significant underestimation of the increased risk associated with the use of tolcapone tablets.
During the premarketing development of tolcapone, two distinct patient populations were studied, patients with end-of-dose wearing-off phenomena and patients with stable responses to levodopa therapy. All patients received concomitant treatment with levodopa preparations, however, and were similar in other clinical aspects. Adverse reactions are shown for these two populations combined.
The most commonly observed adverse reactions in the double-blind, placebo-controlled trials (N=892), with a difference in incidence (tolcapone tablets minus Placebo) of at least 5 % or greater in the 100 mg or 200 mg tolcapone tablets- treated groups compared to placebo, were dyskinesia, nausea, diarrhea, anorexia, sleep disorder, vomiting, urine discoloration, somnolence, hallucination, dystonia, and sweating.
Approximately 16% of the 592 patients who participated in the double-blind, placebo- controlled trials discontinued treatment due to adverse reactions compared to 10% of the 298 patients who received placebo. Diarrhea was by far the most frequent cause of discontinuation (approximately 6% in tolcapone patients vs. 1% on placebo).
Adverse Reaction Incidence in Controlled Clinical Studies:
Table 4 lists treatment emergent adverse reactions that occurred in at least 1% of patients treated with tolcapone participating in the double-blind, placebo-controlled studies and were numerically more common in at least one of the tolcapone groups. In these studies, either tolcapone or placebo was added to levodopa/carbidopa (or benserazide).
The prescriber should be aware that these figures cannot be used to predict the incidence of adverse reactions in the course of usual medical practice where patient characteristics and other factors differ from those that prevailed in the clinical studies. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. However, the cited figures do provide the prescriber with some basis for estimating the relative contribution of drug and nondrug factors to the adverse reactions incidence rate in the population studied.
| Placebo | Tolcapone tid | ||
| N = 298 | 100 mg N = 296 | 200 mg N = 298 | |
| Adverse Reactions | (%) | (%) | (%) |
| Dyskinesia | 20 | 42 | 51 |
| Nausea | 18 | 30 | 35 |
| Sleep Disorder | 18 | 24 | 25 |
| Dystonia | 17 | 19 | 22 |
| Dreaming Excessive | 17 | 21 | 16 |
| Anorexia | 13 | 19 | 23 |
| Cramps Muscle | 17 | 17 | 18 |
| Orthostatic Complaints | 14 | 17 | 17 |
| Somnolence | 13 | 18 | 14 |
| Diarrhea | 8 | 16 | 18 |
| Confusion | 9 | 11 | 10 |
| Dizziness | 10 | 13 | 6 |
| Headache | 7 | 10 | 11 |
| Hallucination | 5 | 8 | 10 |
| Vomiting | 4 | 8 | 10 |
| Constipation | 5 | 6 | 8 |
| Fatigue | 6 | 7 | 3 |
| Upper Respiratory Tract Infection | 3 | 5 | 7 |
| Falling | 4 | 4 | 6 |
| Sweating Increased | 2 | 4 | 7 |
| Urinary Tract Infection | 4 | 5 | 5 |
| Xerostomia | 2 | 5 | 6 |
| Abdominal Pain | 3 | 5 | 6 |
| Syncope | 3 | 4 | 5 |
| Urine Discoloration | 1 | 2 | 7 |
| Dyspepsia | 2 | 4 | 3 |
| Influenza | 2 | 3 | 4 |
| Dyspnea | 2 | 3 | 3 |
| Balance Loss | 2 | 3 | 2 |
| Flatulence | 2 | 2 | 4 |
| Hyperkinesia | 1 | 3 | 2 |
| Chest Pain | 1 | 3 | 1 |
| Hypotension | 1 | 2 | 2 |
| Paresthesia | 2 | 3 | 1 |
| Stiffness | 1 | 2 | 2 |
| Arthritis | 1 | 2 | 1 |
| Chest Discomfort | 1 | 1 | 2 |
| Hypokinesia | 1 | 1 | 3 |
| Micturition Disorder | 1 | 2 | 1 |
| Pain Neck | 1 | 2 | 2 |
| Burning | 0 | 2 | 1 |
| Sinus Congestion | 0 | 2 | 1 |
| Agitation | 0 | 1 | 1 |
| Bleeding Dermal | 0 | 1 | 1 |
| Irritability | 0 | 1 | 1 |
| Mental Deficiency | 0 | 1 | 1 |
| Hyperactivity | 0 | 1 | 1 |
| Malaise | 0 | 1 | 0 |
| Panic Reaction | 0 | 1 | 0 |
| Tumor Skin | 0 | 1 | 0 |
| Cataract | 0 | 1 | 0 |
| Euphoria | 0 | 1 | 0 |
| Fever | 0 | 0 | 1 |
| Alopecia | 0 | 1 | 0 |
| Eye Inflamed | 0 | 1 | 0 |
| Hyertonia | 0 | 0 | 1 |
| Tumor Uterus | 0 | 1 | 0 |
Effects of Gender on Adverse Reactions: Female patients may be more likely to develop somnolence than males.
Other Adverse Events Observed During All Trials in Patients With Parkinson's Disease:
During these trials, all adverse events were recorded by the clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals having adverse events, similar types of adverse events were grouped into a smaller number of standardized categories using COSTART dictionary terminology. These categories are used in the listing below.
All reported events that occurred at least twice (or once for serious or potentially serious events), except those already listed above, trivial events and terms too vague to be meaningful are included, without regard to determination of a causal relationship to tolcapone tablets.
Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in at least 1/100 patients; infrequent adverse events are defined as those occurring in between 1/100 and 1/1000 patients; and rare adverse events are defined as those occurring in fewer than 1/1000 patients.
Nervous System — frequent: depression, hypesthesia, tremor, speech disorder, vertigo, emotional lability; infrequent: neuralgia, amnesia, extrapyramidal syndrome, hostility, libido increased, manic reaction, nervousness, paranoid reaction, cerebral ischemia, cerebrovascular accident, delusions, libido decreased, neuropathy, apathy, choreoathetosis, myoclonus, psychosis, thinking abnormal, twitching; rare: antisocial reaction, delirium, encephalopathy, hemiplegia, meningitis.
Digestive System — frequent: tooth disorder; infrequent: dysphagia, gastrointestinal hemorrhage, gastroenteritis, mouth ulceration, increased salivation, abnormal stools, esophagitis, cholelithiasis, colitis, tongue disorder, rectal disorder; rare: cholecystitis, duodenal ulcer, gastrointestinal carcinoma, stomach atony.
Body as a Whole — frequent: flank pain, accidental injury, abdominal pain, infection; infrequent: hernia, pain, allergic reaction, cellulitis, infection fungal, viral infection, carcinoma, chills, infection bacterial, neoplasm, abscess, face edema; rare: death.
Cardiovascular System — frequent: palpitation; infrequent: hypertension, vasodilation, angina pectoris, heart failure, atrial fibrillation, tachycardia, migraine, aortic stenosis, arrhythmia, arteriospasm, bradycardia, cerebral hemorrhage, coronary artery disorder, heart arrest, myocardial infarct, myocardial ischemia, pulmonary embolus; rare: arteriosclerosis,cardiovascular disorder, pericardial effusion, thrombosis.
Musculoskeletal System — frequent: myalgia; infrequent: tenosynovitis, arthrosis, joint disorder.
Urogenital System — frequent: urinary incontinence, impotence; infrequent: prostatic disorder, dysuria, nocturia, polyuria, urinary retention, urinary tract disorder, hematuria, kidney calculus, prostatic carcinoma, breast neoplasm, oliguria, uterine atony, uterine disorder, vaginitis; rare: bladder calculus, ovarian carcinoma, uterine hemorrhage.
Respiratory System — frequent: bronchitis, pharyngitis; infrequent: cough increased, rhinitis, asthma, epistaxis, hyperventilation, laryngitis, hiccup; rare: apnea, hypoxia, lung edema.
Skin and Appendages — frequent: rash; infrequent: herpes zoster, pruritus, seborrhea, skin discoloration, eczema, erythema multiforme, skin disorder, furunculosis, herpes simplex urticaria.
Special Senses — frequent: tinnitus; infrequent: diplopia, ear pain, eye hemorrhage, eye pain, lacrimation disorder, otitis media, parosmia; rare: glaucoma
Metabolic and Nutritional — infrequent: edema, hypercholesteremia, thirst, dehydration.
Hemic and Lymphatic System — infrequent: anemia; rare: leukemia, thrombocytopenia.
Endocrine System — infrequent: diabetes mellitus.
Unclassified — infrequent: surgical procedure.
DESCRIPTION
Tolcapone tablets, USP is available as tablets containing 100 mg tolcapone, USP.
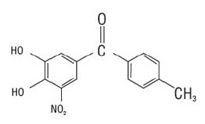
Tolcapone, an inhibitor of catechol-O-methyltransferase (COMT), is used in the treatment of Parkinson's disease as an adjunct to levodopa/carbidopa therapy. It is a yellow, non-hygroscopic, fine powder or fine powder with lumps with a relative molecular mass of 273.24. The chemical name of tolcapone is 3,4-dihydroxy-4'-methyl-5-nitrobenzophenone. Its empirical formula is C 14 H 11 NO 5 and its structural formula is:
Inactive ingredients: Core: lactose monohydrate, microcrystalline cellulose, dibasic calcium phosphate anhydrous, sodium starch glycolate, povidone, talc and magnesium stearate.Film coating: hydroxypropyl methylcellulose, titanium dioxide, talc, ethyl cellulose, triacetin,and sodium lauryl sulfate, with the following dye system: yellow iron oxide.
CLINICAL PHARMACOLOGY
Mechanism of Action:
Tolcapone is a selective and reversible inhibitor of catechol-O-methyltransferase (COMT).
In mammals, COMT is distributed throughout various organs. The highest activities are in the liver and kidney. COMT also occurs in the heart, lung, smooth and skeletal muscles, intestinal tract, reproductive organs, various glands, adipose tissue, skin, blood cells and neuronal tissues, especially in glial cells. COMT catalyzes the transfer of the methyl group of S-adenosyl-Lmethionine to the phenolic group of substrates that contain a catechol structure. Physiological substrates of COMT include dopa, catecholamines (dopamine, norepinephrine, epinephrine) and their hydroxylated metabolites. The function of COMT is the elimination of biologically active catechols and some other hydroxylated metabolites. In the presence of a decarboxylase inhibitor, COMT becomes the major metabolizing enzyme for levodopa catalyzing the metabolism to 3-methoxy-4 hydroxy-L-phenylalanine (3-OMD) in the brain and periphery.
The precise mechanism of action of tolcapone is unknown, but it is believed to be related to its ability to inhibit COMT and alter the plasma pharmacokinetics of levodopa. When tolcapone is given in conjunction with levodopa and an aromatic amino acid decarboxylase inhibitor, such as carbidopa, plasma levels of levodopa are more sustained than after administration of levodopa and an aromatic amino acid decarboxylase inhibitor alone. It is believed that these sustained plasma levels of levodopa result in more constant dopaminergic stimulation in the brain, leading to greater effects on the signs and symptoms of Parkinson's disease in patients as well as increased levodopa adverse effects, sometimes requiring a decrease in the dose of levodopa. Tolcapone enters the CNS to a minimal extent, but has been shown to inhibit central COMT activity in animals.
Pharmacodynamics
COMT Activity in Erythrocytes: Studies in healthy volunteers have shown that tolcapone reversibly inhibits human erythrocyte catechol- O - methyltransferase (COMT) activity after oral administration. The inhibition is closely related to plasma tolcapone concentrations. With a 200-mg single dose of tolcapone, maximum inhibition of erythrocyte COMT activity is on average greater than 80%. During multiple dosing with tolcapone (200 mg tid), erythrocyte COMT inhibition at trough tolcapone blood concentrations is 30% to 45%.
Effect on the Pharmacokinetics of Levodopa and its Metabolites:
When tolcapone is administered together with levodopa/carbidopa, it increases the relative bioavailability (AUC) of levodopa by approximately twofold. This is due to a decrease in levodopa clearance resulting in a prolongation of the terminal elimination half-life of levodopa (from approximately 2 hours to 3.5 hours). In general, the average peak levodopa plasma concentration (C max ) and the time of its occurrence (T max ) are unaffected. The onset of effect occurs after the first administration and is maintained during long-term treatment. Studies in healthy volunteers and Parkinson's disease patients have confirmed that the maximal effect occurs with 100 mg to 200 mg tolcapone. Plasma levels of 3-OMD are markedly and dose-dependently decreased by tolcapone when given with levodopa/carbidopa.
Population pharmacokinetic analyses in patients with Parkinson's disease have shown the same effects of tolcapone on levodopa plasma concentrations that occur in healthy volunteers.
Pharmacokinetics of Tolcapone:
Tolcapone pharmacokinetics are linear over the dose range of 50 mg to 400 mg, independent of levodopa/carbidopa co-administration. The elimination half-life of tolcapone is 2 to 3 hours and there is no significant accumulation. With tid dosing of 100 mg or 200 mg, C max is approximately 3 mcg/mL and 6 mcg/mL, respectively.
Absorption: Tolcapone is rapidly absorbed, with a T max of approximately 2 hours. The absolute bioavailability following oral administration is about 65%. Food given within 1 hour before and 2 hours after dosing of tolcapone decreases the relative bioavailability by 10% to 20% (see DOSAGE AND ADMINISTRATION ).
Distribution: The steady-state volume of distribution of tolcapone is small (9 L). Tolcapone does not distribute widely into tissues due to its high plasma protein binding. The plasma protein binding of tolcapone is >99.9% over the concentration range of 0.32 to 210 mcg/mL. In vitro experiments have shown that tolcapone binds mainly to serum albumin.
Metabolism and Elimination: Tolcapone is almost completely metabolized prior to excretion, with only a very small amount (0.5% of dose) found unchanged in urine. The main metabolic pathway of tolcapone is glucuronidation; the glucuronide conjugate is inactive. In addition, the compound is methylated by COMT to 3- O -methyl-tolcapone. Tolcapone is metabolized to a primary alcohol (hydroxylation of the methyl group), which is subsequently oxidized to the carboxylic acid. In vitro experiments suggest that the oxidation may be catalyzed by cytochrome P450 3A4 and P450 2A6. The reduction to an amine and subsequent N-acetylation occur to a minor extent. After oral administration of a 14 C-labeled dose of tolcapone, 60% of labeled material is excreted in urine and 40% in feces. Tolcapone is a low-extraction-ratio drug (extraction ratio = 0.15) with a moderate systemic clearance of about 7 L/h.
Special Populations:
Tolcapone pharmacokinetics are independent of sex, age, body weight, and race (Japanese, Black and Caucasian). Polymorphic metabolism is unlikely based on the metabolic pathways involved.
Hepatic Impairment: A study in patients with hepatic impairment has shown that moderate non-cirrhotic liver disease had no impact on the pharmacokinetics of tolcapone. In patients with moderate cirrhotic liver disease (Child-Pugh Class B), however, clearance and volume of distribution of unbound tolcapone was reduced by almost 50%. This reduction may increase the average concentration of unbound drug by twofold (see DOSAGE AND ADMINISTRATION ). Tolcapone tablets therapy should not be initiated if the patient exhibits clinical evidence of active liver disease or two SGPT/ALT or SGOT/AST values greater than the upper limit of normal (see BOXED WARNING ).
Renal Impairment: The pharmacokinetics of tolcapone have not been investigated in a specific renal impairment study. However, the relationship of renal function and tolcapone pharmacokinetics has been investigated using population pharmacokinetics during clinical trials. The data of more than 400 patients have confirmed that over a wide range of creatinine clearance values (30 mL/min to 130 mL/min) the pharmacokinetics of tolcapone are unaffected by renal function. This could be explained by the fact that only a negligible amount of unchanged tolcapone (0.5%) is excreted in the urine. The glucuronide conjugate of tolcapone is mainly excreted in the urine but is also excreted in the bile. Accumulation of this stable and inactive metabolite should not present a risk in renally impaired patients with creatinine clearance above 25 mL/min (see DOSAGE AND ADMINISTRATION ). Given the very high protein binding of tolcapone, no significant removal of the drug by hemodialysis would be expected
Drug Interactions: See PRECAUTIONS : Drug Interactions.
Clinical Studies:
The effectiveness of tolcapone tablets as an adjunct to levodopa in the treatment of Parkinson's disease was established in three multicenter randomized controlled trials of 13 to 26 weeks' duration, supported by four 6-week trials whose results were consistent with those of the longer trials. In two of the longer trials, tolcapone was evaluated in patients whose Parkinson's disease was characterized by deterioration in their response to levodopa at the end of a dosing interval (so-called fluctuating patients with wearing-off phenomena). In the remaining trial, tolcapone was evaluated in patients whose response to levodopa was relatively stable (so-called non- fluctuators).
Fluctuating Patients: In two 3-month trials, patients with documented episodes of wearing-off phenomena, despite optimum levodopa therapy, were randomized to receive placebo, tolcapone 100 mg tid or 200 mg tid. The formal double-blind portion of the trial was 3 months long, and the primary outcome was a comparison between treatments in the change from baseline in the amount of time spent "On" (a period of relatively good functioning) and "Off" (a period of relatively poor functioning). Patients recorded periodically, throughout the duration of the trial, the time spent in each of these states.
In addition to the primary outcome, patients were also assessed using sub-parts of the Unified Parkinson's Disease Rating Scale (UPDRS), a frequently used multi-item rating scale intended to evaluate mentation (Part I), activities of daily living (Part II), motor function (Part III), complications of therapy (Part IV), and disease staging (Parts V and VI); an Investigator's Global Assessment of Change (IGA), a subjective scale designed to assess global functioning in 5 areas of Parkinson's disease; the Sickness Impact Profile (SIP), a multi-item scale in 12 domains designed to assess the patient's functioning in multiple areas; and the change in daily levodopa/carbidopa dose.
In one of the studies, 202 patients were randomized in 11 centers in the United States and Canada. In this trial, all patients were receiving concomitant levodopa and carbidopa. In the second trial, 177 patients were randomized in 24 centers in Europe. In this trial, all patients were receiving concomitant levodopa and benserazide.
The following tables display the results of these 2 trials:
• Compared to placebo. Nominal p values are not adjusted for multiple comparisons. | |||
•• Hours “Off” or “On” are based on the percent of waking day “Off” or “On”, assuming a 16-hour waking day. | |||
| Primary Measure | |||
| Baseline ( hrs ) | Change from Baseline at Month 3 ( hrs ) | p - value • | |
| Hours of Wake Time “ Off ” •• | |||
| Placebo | 6.2 | -1.2 | --- |
| 100 mg tid | 6.4 | -2.0 | 0.169 |
| 200 mg tid | 5.9 | -3.0 | <0.001 |
| Hours of Wake Time “ On ” •• | |||
| Placebo | 8.7 | 1.4 | --- |
| 100 mg tid | 8.1 | 2.0 | 0.267 |
| 200 mg tid | 9.1 | 2.9 | <0.008 |
| Secondary Measure | |||
| Baseline | Change from Baseline at Month 3 | p - value • | |
| Levodopa Total Daily Dose ( mg ) | |||
| Placebo | 948 | 16 | --- |
| 100 mg tid | 788 | -166 | <0.001 |
| 200 mg tid | 865 | -207 | <0.001 |
| Global ( overall ) % Improved | |||
| Placebo | --- | 42 | --- |
| 100 mg tid | --- | 71 | <0.001 |
| 200 mg tid | --- | 91 | <0.001 |
| UPDRS Motor | |||
| Placebo | 19.5 | -0.4 | --- |
| 100 mg tid | 17.6 | -1.9 | 0.217 |
| 200 mg tid | 20.6 | -2.0 | 0.210 |
| UPDRS ADL | |||
| Placebo | 7.5 | -0.3 | --- |
| 100 mg tid | 7.7 | -0.8 | 0.487 |
| 200 mg tid | 8.3 | 0.2 | 0.412 |
| SIP ( total ) | |||
| Placebo | 14.7 | -2.2 | --- |
| 100 mg tid | 14.9 | -0.4 | 0.210 |
| 200 mg tid | 17.6 | -0.3 | 0.216 |
• Compared to placebo. Nominal p values are not adjusted for multiple comparisons. | |||
•• Hours “Off” or “On” are based on the percent of waking day “Off” or “On”, assuming a 16- hour waking day. | |||
| Primary Measure | |||
| Baseline ( hrs ) | Change from Baseline at Month 3 ( hrs ) | p - value • | |
| Hours of Wake Time “ Off ” •• | |||
| Placebo | 6.1 | -0.7 | --- |
| 100 mg tid | 6.5 | -2.0 | 0.008 |
| 200 mg tid | 6.0 | -1.6 | 0.081 |
| Hours of Wake Time “ On ” •• | |||
| Placebo | 8.5 | -0.1 | --- |
| 100 mg tid | 8.1 | 1.7 | 0.003 |
| 200 mg tid | 8.4 | 1.7 | 0.003 |
| Secondary Measure | |||
| Baseline | Change from Baseline at Month 3 | p - value • | |
| Levodopa Total Daily Dose ( mg ) | |||
| Placebo | 660 | -29 | --- |
| 100 mg tid | 667 | -109 | 0.025 |
| 200 mg tid | 675 | -122 | 0.010 |
| Global ( overall ) % Improved | |||
| Placebo | --- | 37 | --- |
| 100 mg tid | --- | 70 | 0.003 |
| 200 mg tid | --- | 78 | <0.001 |
| UPDRS Motor | |||
| Placebo | 24.0 | -2.1 | --- |
| 100 mg tid | 22.4 | -4.2 | 0.163 |
| 200 mg tid | 22.4 | -6.5 | 0.004 |
| UPDRS ADL | |||
| Placebo | 7.9 | -0.5 | --- |
| 100 mg tid | 7.5 | -0.9 | 0.408 |
| 200 mg tid | 7.7 | -1.3 | 0.097 |
| SIP ( total ) | |||
| Placebo | 21.6 | -0.9 | --- |
| 100 mg tid | 16.6 | -1.9 | 0.419 |
| 200 mg tid | 18.4 | -4.2 | 0.011 |
Effects on "Off" time and levodopa dose did not differ by age or sex.
Non-fluctuating Patients: In this study, 298 patients with idiopathic Parkinson's disease on stable doses of levodopa/carbidopa who were not experiencing wearing-off phenomena were randomized to placebo, tolcapone 100 mg tid, or tolcapone 200 mg tid for 6 months at 20 centers in the United States and Canada. The primary measure of effectiveness was the Activities of Daily Living portion (Subscale II) of the UPDRS. In addition, the change in daily levodopa dose, other subscales of the UPDRS, and the SIP were assessed as secondary measures. The results are displayed in the following table
• Compared to placebo. Nominal p values are not adjusted for multiple comparisons. | |||
| Primary Measure | |||
| Baseline ( hrs ) | Change from Baseline at Month 6 | p - value • | |
| UPDRS ADL | |||
| Placebo | 8.5 | 0.1 | --- |
| 100 mg tid | 7.5 | -1.4 | <0.001 |
| 200 mg tid | 7.9 | -1.6 | <0.001 |
| Secondary Measure | |||
| Baseline | Change from Baseline at Month 6 | p - value • | |
| Levodopa Total Daily Dose ( mg ) | |||
| Placebo | 364 | 47 | --- |
| 100 mg tid | 370 | -21 | <0.001 |
| 200 mg tid | 381 | -32 | <0.001 |
| UPDRS Motor | |||
| Placebo | 19.7 | 0.1 | --- |
| 100 mg tid | 17.3 | -2.0 | 0.018 |
| 200 mg tid | 16.0 | -2.3 | 0.008 |
| SIP ( total ) | |||
| Placebo | 6.9 | 0.4 | --- |
| 100 mg tid | 7.3 | -0.9 | 0.044 |
| 200 mg tid | 7.3 | -0.7 | 0.078 |
| Percent of Patients who Developed Fluctuations | |||
| Placebo | --- | 26 | --- |
| 100 mg tid | --- | 19 | 0.297 |
| 200 mg tid | --- | 14 | 0.047 |
Effects on Activities of Daily Living did not differ by age or sex.
HOW SUPPLIED
Tolcapone tablets USP, 100 mg is supplied as pale yellow to yellow color biconvex, oval shaped film-coated, unscored tablets, debossed with 'RA' on one side and '10' on other side.
Tolcapone tablets USP, 100 mg: bottles of 90 (NDC 50742-193-90).
Storage: Store in tight containers at 20°C-25°C (68°F-77°F); excursions permitted to 15°C- 30°C (59°F-86°F) [See USP Controlled Room Temperature].
| PATIENT ACKNOWLEDGMENT OF RISKS ASSOCIATED WITH TOLCAPONE TABLETS TREATMENT |
| The following is important information that patients should know about tolcapone tablets. |
| ● Tolcapone tablets should not be used until you and your doctor (insert physician name here: ______________________________________) have had a complete discussion about the risks and benefits associated with the use of tolcapone tablets. ● Reports of potentially life-threatening cases of severe hepatocellular injury, including fulminant liver failure resulting in death, have been reported in association with use of tolcapone tablets. ● There are no laboratory tests that will predict in advance which patients are at an increased risk for liver failure or death from liver failure. ● Patients should have the recommended liver blood tests before treatment with tolcapone tablets is begun and periodically for the first 6 months of therapy. After the first six months, periodic liver blood tests should be performed as directed by your physician. If the dose of tolcapone tablets is to be increased, the liver blood tests should be checked before increasing the dose and repeated periodically as described earlier. Liver blood tests may help detect if liver failure has occurred but they may do so only after significant damage, that may not go away, has already occurred. ● Patients must immediately report any unusual symptoms to their physician and be especially aware of persistent nausea, fatigue, lethargy, decreased appetite, jaundice (yellowing of skin or the whites of the eyes), dark urine, itchiness or right- sided abdominal pain. |
| The above points of information, possibly along with other information, have been explained to me and I have been able to ask my physician questions and discuss risks and benefits associated with tolcapone tablets treatment. |
| Patient or Patient Caregiver Signature: ___________________________________________________ |
| Date: ________________________________ |
| NOTETOPHYSICIAN:It is strongly recommended that you retain a signed copy of this form with the patient's medical records. |
| SUPPLYOFPATIENTACKNOWLEDGEMENTFORMS: |
| A supply of Patient Acknowledgement forms is available, free of charge, from your local Ingenus Pharmaceuticals representative, or by calling at 1-877-748-1970. Permission to use the above Patient Acknowledgement form by photocopy reproduction is also hereby granted by Ingenus Pharmaceuticals, LLC. U.S.A. |
Rx only
Manufactured for:
Ingenus Pharmaceuticals, LLC
Orlando, FL 32839-6408
Made in India
Rev: 08/2018