Get your patient on Xromi - Hydroxyurea solution (Hydroxyurea)
Xromi - Hydroxyurea solution prescribing information
WARNING: MYELOSUPPRESSION and MALIGNANCIES
- Myelosuppression: XROMI may cause severe myelosuppression. Do not give if bone marrow function is markedly depressed. Monitor blood counts at baseline and throughout treatment. Interrupt treatment and reduce dose as necessary [see Warnings and Precautions(5.1) ] .
- Malignancies: Hydroxyurea is carcinogenic. Advise sun protection and monitor patients for malignancies [see Warnings and Precautions (5.3) ] .
INDICATIONS AND USAGE
XROMI is indicated to reduce the frequency of painful crises and reduce the need for blood transfusions in pediatric patients aged 6 months of age and older with sickle cell anemia with recurrent moderate to severe painful crises.
DOSAGE AND ADMINISTRATION
- Initial dose: 15 mg/kg orally once daily. Monitor the patient’s blood count every two weeks. (2.1)
- The dose may be increased by 5 mg/kg/day every 8 to 12 weeks until a maximum tolerated dose or 35 mg/kg/day is reached if blood counts are in an acceptable range. (2.1)
- The dose is not increased if blood counts are below the acceptable range and toxic. Discontinue XROMI until hematologic recovery if blood counts are considered toxic. Treatment may be resumed after reducing the dose by 2.5mg/kg/day to 5mg/kg/day from the dose associated with hematological toxicity. (2.1)
- Renal impairment: Reduce the dose of XROMI by 50% in patients with creatinine clearance less than 60 mL/min. (2.2 , 8.6 , 12.3)
Recommended Dosage
The recommended XROMI dosage in pediatric patients aged 6 months and older is described in Table 1.
Dosing Regimen | Dose | Dose modification criteria | Monitoring parameters |
Initial Recommended dosing | 15 mg/kg/day (rounded to nearest 10 mg) orally as a single dose once daily based on the patient’s actual body weight. | Monitor the patient’s complete blood count (CBC) with differential and reticulocyte count every 2 weeks while adjusting dosage [see Warnings and Precautions (5.1) ] . | |
Dosing Adjustment Based on Blood Counts in the acceptable range | Increase dose 5 mg/kg/day every 8 to 12 weeks. Maximal dose: 35 mg/kg/day.• •Maximal dose is the highest dose that does not produce toxic blood counts over 24 consecutive weeks. | Increase dose only if blood counts are in an acceptable range. Do not increase if myelosuppression occurs. | Target Blood Counts Absolute neutrophil count (ANC) 1 to 3 x 10 9 /L and platelets at least 80 x 10 9 /L |
Dosing Adjustment Based on Blood Counts below acceptable range | Do not increase dose. | If blood counts are considered toxic, discontinue XROMI until hematologic recovery. | Blood Counts Toxic Range
|
Dosing after Hematologic Recovery | If hematologic toxicity resolved within 1 week, restart at the same XROMI dose. If hematologic toxicity persisted for more than 1 week or occurred twice in a 3 month period, reduce dose by 5 mg/kg/day. | Once a stable dose is established, monitor CBC with differential and reticulocyte count every 4 weeks for 2 months and then as clinically indicated. |
Caregivers must be able to follow directions regarding drug administration and their monitoring and care.
If a dose of XROMI is missed at the scheduled time, the patient should take the missed dose as soon as possible once it is noticed, but only on the same day. If this is not possible, the patient should skip the dose and continue with the next dose as prescribed.
The patient should not take two doses to make up for a missed dose.
Fetal hemoglobin (HbF) levels may be used to evaluate the efficacy of XROMI in clinical use. Obtain HbF levels every three to four months. Monitor for an increase in HbF of at least two-fold over the baseline value.
XROMI causes macrocytosis, which may mask the incidental development of folic acid deficiency. Prophylactic administration of folic acid is recommended.
Administration Instructions
XROMI is for oral use. See Instructions for Use for details on preparation and administration of XROMI for oral solution. Do not shake. Two oral dosing syringes (one oral dosing syringe graduated to 3 mL and one oral dosing syringe graduated to 10 mL) are provided for accurate measurement of the prescribed dose of the oral solution. It is recommended that the healthcare professional advises the caregiver which oral dosing syringe to use to ensure that the correct volume is administered. The smaller 3 mL oral dosing syringe, marked from 0.5 mL to 3 mL, is for measuring doses of less than or equal to 3 mL. This oral dosing syringe should be recommended for doses less than or equal to 3 mL (each graduation of 0.1 mL contains 10 mg of hydroxyurea). The larger 10 mL oral dosing syringe, marked 1 mL to 10 mL, is for measuring doses of more than 3 mL. This oral dosing syringe should be recommended for doses greater than 3 mL (each graduation of 0.25 mL contains 25 mg of hydroxyurea). XROMI may be taken with or after meals at any time of the day but caregivers should standardize the method of administration and time of day. XROMI is a hazardous drug. Follow applicable special handling and disposal procedures [see Reference (15) ] .
Dosage Modifications in Renal Impairment
Reduce the dose of XROMI by 50% in patients with creatinine clearance of less than 60 mL/min or with end-stage renal disease (ESRD) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3) ] . Creatinine clearance were obtained using 24-hour urine collection.
| Creatinine Clearance (mL/min) | Recommended XROMI Initial Dose |
| Greater than or equal to 60 | 15 mg/kg once daily |
| Less than 60 or ESRD• | 7.5 mg/kg once daily |
| • On dialysis days, administer XROMI to patients with ESRD following hemodialysis |
Monitor the hematologic parameters closely in these patients.
DOSAGE FORMS AND STRENGTHS
Oral solution: 100 mg/mL clear colorless to pale yellow liquid in a multiple-dose amber bottle.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary XROMI can cause fetal harm based on findings from animal studies and the drug’s mechanism of action [see Clinical Pharmacology (12.1) ]. There are no data with XROMI use in pregnant women to inform a drug-associated risk. In animal reproduction studies, administration of hydroxyurea to pregnant rats and rabbits during organogenesis produced embryotoxic and teratogenic effects at doses 0.8 times and 0.3 times, respectively, the maximum recommended human daily dose on a mg/m 2 basis (see Data ). Advise women of the potential risk to a fetus and to avoid becoming pregnant while being treated with XROMI. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15%–20%, respectively.
Data Animal Data Hydroxyurea has been demonstrated to be a potent teratogen in a wide variety of animal models, including mice, hamsters, cats, miniature swine, dogs, and monkeys at doses within 1-fold of the human dose given on a mg/m 2 basis. Hydroxyurea is embryotoxic and causes fetal malformations (partially ossified cranial bones, absence of eye sockets, hydrocephaly, bipartite sternebrae, missing lumbar vertebrae) at 180 mg/kg/day (about 0.8 times the maximum recommended human daily dose on a mg/m 2 basis) in rats and at 30 mg/kg/day (about 0.3 times the maximum recommended human daily dose on a mg/m 2 basis) in rabbits. Embryotoxicity was characterized by decreased fetal viability, reduced live litter sizes, and developmental delays. Hydroxyurea crosses the placenta. Single doses of ≥375 mg/kg (about 1.7 times the maximum recommended human daily dose on a mg/m 2 basis) to rats caused growth retardation and impaired learning ability.
Lactation
Risk Summary It is not known whether XROMI is excreted in human milk, the effects of XROMI on the breastfed child, or the effects of XROMI on milk production. Because of the potential for serious adverse reactions in a breastfed child from XROMI, including carcinogenicity, advise patients not to breastfeed during treatment with XROMI.
Females and Males of Reproductive Potential
Pregnancy Testing Verify the pregnancy status of females of reproductive potential prior to initiating XROMI therapy. Contraception Females XROMI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) ]. Advise females of reproductive potential to use effective contraception during and after treatment with XROMI for at least 6 months after therapy. Advise females to immediately report pregnancy. Males XROMI may damage spermatozoa and testicular tissue, resulting in possible genetic abnormalities. Males with female sexual partners of reproductive potential should use effective contraception during and after treatment with XROMI for at least 1 year after therapy [see Nonclinical Toxicology (13.1) ]. Infertility Males Based on findings in animals and humans, male fertility may be compromised by treatment with XROMI. Azoospermia or oligospermia, sometimes reversible, has been observed in men. Inform male patients about the possibility of sperm conservation before the start of therapy [see Nonclinical Toxicology (13.1) ].
Pediatric Use
The safety and effectiveness of XROMI have been established in pediatric patients aged 6 months and older with sickle cell anemia with recurrent moderate to severe painful crises. Use of XROMI in these age groups is supported by evidence from a pharmacokinetic, efficacy and safety study, in which 32 pediatric patients ages 6 months to <18 years were enrolled. Among the 32 pediatric patients treated with XROMI, 6 were infants (6 months – 2 years), 16 children (2-6 years) and 10 were adolescents (6-18 years) [see Clinical Studies (14) ]. Continuous follow-up of the growth of treated children is recommended.
Renal Impairment
The exposure to XROMI is higher in patients with creatinine clearance of less than 60 mL/min. Reduce dosage and closely monitor the hematologic parameters when XROMI is to be administered to these patients [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3) ].
Hepatic Impairment
There are no data that support specific guidance for dosage adjustment in patients with hepatic impairment. Close monitoring of hematologic parameters is advised in these patients.
CONTRAINDICATIONS
XROMI is contraindicated in patients:
- who have demonstrated a previous hypersensitivity to hydroxyurea or any other component of its formulation. [see Adverse Reactions (6) ] .
WARNINGS AND PRECAUTIONS
- Hemolytic anemia: Monitor blood counts throughout treatment. If hemolysis persists, discontinue XROMI. (5.2 )
- Embryo-Fetal toxicity: Can cause fetal harm. Advise of potential risk to a fetus and use of effective contraception. (5.4 , 8.1 , 8.3)
- Vasculitic toxicities: Institute treatment and discontinue XROMI if this occurs. (5.5)
- Live Vaccinations: Avoid live vaccine use in a patient taking XROMI. (5.6)
- Risks with concomitant use of antiretroviral drugs: Pancreatitis, hepatotoxicity, and neuropathy have occurred. Monitor for signs and symptoms in patients with HIV infection using antiretroviral drugs; discontinue XROMI and implement treatment. (5.7)
Myelosuppression
Hydroxyurea causes severe myelosuppression. Do not initiate treatment with XROMI in patients if bone marrow function is markedly depressed. Bone marrow suppression may occur, and leukopenia is generally its first and most common manifestation. Thrombocytopenia and anemia occur less often and are seldom seen without a preceding leukopenia.
Some patients, treated at the recommended initial dose of 15 mg/kg/day, have experienced severe or life-threatening myelosuppression.
Evaluate hematologic status (CBC, reticulocyte count) prior to and every 2 weeks during dose-escalation period of XROMI treatment. Once a stable dose of XROMI is achieved, monitor every 4 weeks. Provide supportive care and modify dose or discontinue XROMI as needed. Recovery from myelosuppression is usually rapid when therapy is interrupted. [see Dosage and Administration (2.1) ] .
5.2 Hemolytic Anemia
Cases of hemolytic anemia in patients treated with hydroxyurea for myeloproliferative diseases have been reported [see Adverse Reactions (6.1) ] . Patients who develop acute jaundice or hematuria in the presence of persistent or worsening of anemia should have laboratory tests evaluated for hemolysis (e.g., measurement of serum lactate dehydrogenase, haptoglobin, reticulocyte, unconjugated bilirubin levels, urinalysis, and direct and indirect antiglobulin [Coombs] tests). In the setting of confirmed diagnosis of hemolytic anemia and in the absence of other causes, discontinue XROMI.
Malignancies
Hydroxyurea is a human carcinogen. In patients receiving long-term hydroxyurea for myeloproliferative disorders (a condition for which XROMI is not approved), secondary leukemia has been reported.
Secondary leukemia has also been reported in patients treated with long-term hydroxyurea for sickle cell anemia. Leukemia has also been reported in patients with sickle cell anemia and no prior history of treatment with hydroxyurea.
All patients using XROMI should be followed up on a long-term basis with regular blood counts to detect development of leukemia.
Skin cancer has also been reported in patients receiving long-term hydroxyurea. Advise protection from sun exposure and monitor for the development of secondary malignancies.
Embryo-Fetal Toxicity with Unapproved Use in Adolescents and Adults
Based on the mechanism of action and findings in animals, XROMI can cause fetal harm when administered to a pregnant woman. Hydroxyurea was embryotoxic and teratogenic in rats and rabbits at doses 0.8 times and 0.3 times, respectively, the maximum recommended human daily dose on a mg/m 2 basis.
Vasculitic Toxicities
Cutaneous vasculitic toxicities, including vasculitic ulcerations and gangrene, have occurred in patients with myeloproliferative disorders during therapy with hydroxyurea. These vasculitic toxicities were reported most often in patients with a history of, or currently receiving, interferon therapy. If cutaneous vasculitic ulcers occur, institute treatment and discontinue XROMI.
Live Vaccinations
Avoid use of live vaccines in patients taking XROMI. Concomitant use of XROMI with a live virus vaccine may potentiate the replication of the virus and/or may increase the adverse reaction of the vaccine because normal defense mechanisms may be suppressed by XROMI. Vaccination with live vaccines in a patient receiving XROMI may result in severe infection [see Drug Interactions (7.2) ] . Patient’s antibody response to vaccines may be decreased. Consider consultation with a specialist.
Risks with Concomitant Use of Antiretroviral Drugs
Pancreatitis, hepatotoxicity, and peripheral neuropathy have occurred when hydroxyurea was administered concomitantly with antiretroviral drugs, including didanosine and stavudine [see Drug Interactions (7.1) ] .
Macrocytosis
XROMI may cause macrocytosis, which is self-limiting, and is often seen early in the course of treatment. The morphologic change resembles pernicious anemia, but is not related to vitamin B 12 or folic acid deficiency. This may mask the diagnosis of pernicious anemia. Prophylactic administration of folic acid is recommended.
Pulmonary Toxicity
Interstitial lung disease including pulmonary fibrosis, lung infiltration, pneumonitis, and alveolitis/allergic alveolitis (including fatal cases) have been reported in patients treated for myeloproliferative neoplasm. Safety and effectiveness have not been established for the use of XROMI in the treatment of myeloproliferative neoplasms and the use is not approved by the FDA. Monitor patients developing pyrexia, cough, dyspnea, or other respiratory symptoms frequently, investigate and treat promptly. Discontinue XROMI and manage with corticosteroids [see Adverse Reactions (6.1) ] .
Laboratory Test Interference
Interference with uric acid, urea, or lactic acid assays is possible, rendering falsely elevated results of these in patients treated with hydroxyurea [see Drug Interactions (7.2) ] .
Hydroxyurea may falsely elevate sensor glucose results from certain continuous glucose monitoring (CGM) systems and may lead to hypoglycemia if sensor glucose results are relied upon to dose insulin.
If a patient using a CGM is to be prescribed hydroxyurea, consult with the CGM prescriber about alternative glucose monitoring methods [see Drug Interactions (7.2) ] .
ADVERSE REACTIONS
The following clinically significant adverse reactions are described in detail in other labeling sections:
- Myelosuppression [see Warnings and Precautions (5.1) ]
- Hemolytic anemia [see Warnings and Precautions (5.2) ]
- Malignancies [see Warnings and Precautions (5.3) ]
- Vasculitic toxicities [see Warnings and Precautions (5.5) ]
- Live vaccinations [see Warnings and Precautions (5.6)
- Risks with concomitant use of antiretroviral drugs [see Warnings and Precautions (5.7) ]
- Macrocytosis [see Warnings and Precautions (5.8) ]
- Pulmonary toxicity [see Warnings and Precautions (5.9) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of XROMI was evaluated in 32 pediatric patients aged 10 months -16 years with sickle cell anemia in a single-arm, open-label, prospective, multi-center, pharmacokinetic, safety and efficacy study (HUPK study). Only adverse reactions associated with the use of XROMI in pediatric patients aged 10 months to less than 2 years are presented.
The most frequently reported adverse reactions in HUPK study (<33%) were neutropenia, and thrombocytopenia [see Table 3].
| 10 Months–<2 Years | 2 –<6 Years | 6 –<18 Years | Overall | |
| Body System Adverse Reactions | (n=6)% | (n=16)% | (n=10)% | (n=32)% |
| Neutropenia | 3 (50) | 1 (6) | 0 | 4 (13) |
| Thrombocytopenia | 2 (33) | 1 (6) | 0 | 3 (9) |
| Diarrhea | 1 (17) | 0 | 0 | 1 (3) |
| GGT Increased | 0 | 0 | 1(10) | 1 (3) |
| Absolute Reticulocyte Count Decreased | 1 (17) | 0 | 0 | 1 (3) |
| Alopecia | 0 | 1 (6) | 0 | 1 (3) |
| Nail Discolouration | 0 | 0 | 1 (10) | 1 (3) |
| Rash | 0 | 0 | 1 (10) | 1 (3) |
| Rash Popular | 0 | 1 (6) | 1 (10) | 2 (6) |
Post marketing Experience
The following adverse reactions have been identified during post-approval use of hydroxyurea. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency.
- Reproductive System and Breast disorders: azoospermia, and oligospermia
- Gastrointestinal disorders: stomatitis, nausea, vomiting, diarrhea, and constipation
- Metabolism and Nutrition disorders: anorexia
- Skin and subcutaneous tissue disorders: maculopapular rash, skin ulceration, cutaneous lupus erythematosus, dermatomyositis-like skin changes, peripheral and facial erythema, hyperpigmentation, nail hyperpigmentation, atrophy of skin and nails, scaling, violet papules, and alopecia
- Renal and urinary disorders: dysuria, elevations in serum uric acid, blood urea nitrogen (BUN), and creatinine levels
- Nervous system disorders: headache, dizziness, drowsiness, disorientation, hallucinations, and convulsions
- General disorders: fever, chills, malaise, edema, and asthenia
- Hepatobiliary disorders: elevation of hepatic enzymes, cholestasis, and hepatitis
- Respiratory disorders: diffuse pulmonary infiltrates, dyspnea, and pulmonary fibrosis, interstitial lung disease, pneumonitis, alveolitis, allergic alveolitis and cough
- Immune disorders: systemic lupus erythematosus
- Hypersensitivity: Drug-induced fever (pyrexia) (>39°C, >102°F) requiring hospitalization has been reported concurrently with gastrointestinal, pulmonary, musculoskeletal, hepatobiliary, dermatological or cardiovascular manifestations. Onset typically occurred within 6 weeks of initiation and resolved upon discontinuation of hydroxyurea. Upon re-administration fever reoccurred typically within 24 hours.
- Blood and lymphatic system disorders: hemolytic anemia
DRUG INTERACTIONS
Increased Toxicity with Concomitant Use of Antiretroviral Drugs
Pancreatitis In patients with HIV infection during therapy with hydroxyurea and didanosine, with or without stavudine, fatal and nonfatal cases of pancreatitis have occurred. Hydroxyurea is not indicated for the treatment of HIV infection; however, if patients with HIV infection are treated with hydroxyurea, and in particular, in combination with didanosine and/or stavudine, close monitoring for signs and symptoms of pancreatitis is recommended. Permanently discontinue therapy with hydroxyurea in patients who develop signs and symptoms of pancreatitis.
Hepatotoxicity Hepatotoxicity and hepatic failure resulting in death have been reported during postmarketing surveillance in patients with HIV infection treated with hydroxyurea and other antiretroviral drugs. Fatal hepatic events were reported most often in patients treated with the combination of hydroxyurea, didanosine, and stavudine. Avoid this combination.
Peripheral Neuropathy Peripheral neuropathy, which was severe in some cases, has been reported in patients with HIV infection receiving hydroxyurea in combination with antiretroviral drugs, including didanosine, with or without stavudine.
Laboratory Test Interference
Interference with Uric Acid, Urea, or Lactic Acid Assays
Studies have shown that there is an analytical interference of hydroxyurea with the enzymes (urease, uricase, and lactate dehydrogenase) used in the determination of urea, uric acid, and lactic acid, rendering falsely elevated results of these in patients treated with hydroxyurea.
Interference with Continuous Glucose Monitoring Systems Hydroxyurea may falsely elevate sensor glucose results from certain continuous glucose monitoring (CGM) systems and may lead to hypoglycemia if sensor glucose results are relied upon to dose insulin.
If a patient using a CGM is to be prescribed hydroxyurea, consult with the CGM prescriber about alternative glucose monitoring methods.
DESCRIPTION
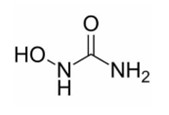
XROMI (hydroxyurea) is available for oral use as oral solution containing 100 mg/mL hydroxyurea. Inactive ingredients include methyl parahydroxybenzoate, purified water, sodium hydroxide, strawberry flavor, sucralose and xanthan gum. Hydroxyurea is a white to off-white crystalline powder. It is hygroscopic and freely soluble in water, but practically insoluble in alcohol. The empirical formula is CH 4 N 2 O 2 and it has a molecular weight of 76.05. Its structural formula is:
CLINICAL PHARMACOLOGY
Mechanism of Action
The precise mechanism by which hydroxyurea produces its cytotoxic and cytoreductive effects is not known. However, various studies support the hypothesis that hydroxyurea causes an immediate inhibition of DNA synthesis by acting as a ribonucleotide reductase inhibitor, without interfering with the synthesis of ribonucleic acid or of protein. The mechanisms by which XROMI produces its beneficial effects in patients with sickle cell anemia are uncertain. Known pharmacologic effects of XROMI that may contribute to its beneficial effects include increasing HbF levels in red blood cells (RBCs), decreasing neutrophils, increasing the water content of RBCs, increasing deformability of sickled cells, and altering the adhesion of RBCs to endothelium.
Pharmacodynamics
In pediatric patients treated with hydroxyurea for up to 15 months, the ratio of fetal hemoglobin to total hemoglobin was 25%, 23.6% and 11.2% at the end of the study, representing a change from baseline increase of 9%, 148% and 167% in patients aged 6 months to <2 years, 2 to <6 years and 6 to <18 years, respectively.
Pharmacokinetics
Absorption Following oral administration of XROMI, hydroxyurea reaches peak plasma concentrations in 0 to 2 hours. Mean peak plasma concentrations and AUCs increase more than proportionally with increase of dose.
Effect of Food There are no data on the effect of food on the absorption of hydroxyurea.
Distribution Hydroxyurea distributes throughout the body with a volume of distribution approximating total body water. Hydroxyurea concentrates in leukocytes and erythrocytes.
Elimination Metabolism Up to 60% of an oral dose undergoes conversion through saturable hepatic metabolism and a minor pathway of degradation by urease found in intestinal bacteria.
Excretion In patients with sickle cell anemia, the mean cumulative urinary recovery of hydroxyurea was about 40% of the administered dose.
Specific Populations Renal Impairment The effect of renal impairment on the pharmacokinetics of hydroxyurea was assessed in adult patients with sickle cell anemia and renal impairment. Patients with normal renal function (creatinine clearance [CrCl] >80 mL/min), mild (CrCl 50-80 mL/min), moderate (CrCl =30-<50 mL/min), or severe (<30 mL/min) renal impairment received a single oral dose of 15 mg/kg hydroxyurea. Creatinine clearance values were obtained using 24-hour urine collections. Patients with ESRD received two doses of 15 mg/kg separated by 7 days; the first was given following a 4-hour hemodialysis session, the second prior to hemodialysis. The exposure to hydroxyurea (mean AUC) in patients with CrCl <60 mL/min and those with ESRD was 64% higher than in patients with normal renal function (CrCl >60 mL/min). [see Dosage and Administration (2.2) and Use in Specific Populations (8.6) ].
Pediatric Patients The model estimated steady state exposures (AUC) in pediatric patients receiving a daily dose of 15 mg/kg is lower in patients aged 6 months to <2 years and 2 to <6 years by 40% and 28% compared to adults receiving the same dose while the exposures in 6 to <18 year old patients are similar to adult exposures (XROMI is not approved for use in adults).
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Conventional long-term studies to evaluate the carcinogenic potential of hydroxyurea have not been performed. However, intraperitoneal administration of 125 to 250 mg/kg hydroxyurea (about 0.6-1.2 times the maximum recommended human oral daily dose on a mg/m 2 basis) thrice weekly for 6 months to female rats increased the incidence of mammary tumors in rats surviving to 18 months compared to control. Hydroxyurea is mutagenic in vitro to bacteria, fungi, protozoa, and mammalian cells. Hydroxyurea is clastogenic in vitro (hamster cells, human lymphoblasts) and in vivo (SCE assay in rodents, mouse micronucleus assay). Hydroxyurea causes the transformation of rodent embryo cells to a tumorigenic phenotype [see Warnings and Precautions (5.3 , 5.4) ] . Hydroxyurea administered to male rats at 60 mg/kg /day (about 0.3 times the maximum recommended human daily dose on a mg/m 2 basis) produced testicular atrophy, decreased spermatogenesis and significantly reduced their ability to impregnate females [see Use in Specific Populations (8.3) ] .
CLINICAL STUDIES
The effectiveness of XROMI has been established for the indication, “to reduce the frequency of painful crises and to reduce the need for blood transfusions in pediatric patients aged 6 months and older with sickle cell anemia with recurrent moderate to severe painful crises” based on an adequate and well-controlled study of hydroxyurea capsules in adult patients with sickle cell anemia with recurrent moderate to severe pain crises and additional pharmacokinetic data from a single-arm, open-label study of XROMI in pediatric patients aged 10 months to less than 18 years with sickle cell anemia, who were treatment naïve or had not received hydroxyurea in the 6 months prior to enrollment: HUPK study [see Adverse Reactions (6.1) ].
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
XROMI (hydroxyurea) 100 mg/mL is a colorless to pale yellow liquid supplied in an Amber type III glass bottle with tamper evident child-resistant closure (HDPE with expanded polyethylene liner) containing 148 mL of oral solution. Each carton NDC 62484-0015-5 contains 1 bottle XROMI NDC 62484-0015-4, an LDPE bottle adaptor and 2 oral dosing syringes (one oral dosing syringe graduated to 3 mL and one oral dosing syringe graduated to 10 mL).
Storage
Store XROMI between 2°C to 8°C (35°F to 46°F) excursions permitted to 25°C (77°F) for up to 72 hours. Keep the bottle tightly closed to protect the integrity of the product and minimize the risk of accidental spillage. Do not freeze. Use within 12 weeks after initially opening the bottle. Discard unused XROMI remaining after 12 weeks of first opening the bottle.
Handling and Disposal
XROMI is a hazardous drug. Follow applicable special handling and disposal procedures [see References (15) ] . Anyone handling XROMI should wash their hands before and after administering a dose. To decrease the risk of exposure, parents and caregivers should wear disposable gloves when handling XROMI. To minimize air bubbles, the bottle should not be shaken prior to dosing. XROMI contact with skin or mucous membrane must be avoided. If XROMI comes into contact with skin or mucosa, it should be washed immediately and thoroughly with soap and water. Spillages must be wiped immediately with a damp disposable towel and discarded in a closed container, such as a plastic bag. The spill areas should be cleaned three times using a detergent solution followed by clean water. If contact with XROMI occurs in the eye(s), flush the eye(s) thoroughly with water or isotonic eyewash designated for that purpose for at least 15 minutes. Parents / care givers should be advised to keep hydroxyurea out of the sight and reach of children. Accidental ingestion can be lethal for children. Oral dosing syringes should be rinsed and washed with cold or warm water and dried completely before the next use. Store oral dosing syringes in a hygienic place with the medicine.
| INSTRUCTIONS FOR USE XROMI ® (ex-ro-mee) hydroxyurea) oral solution |
Read these Instructions for Use before your child starts taking XROMI, and each time you get a refill. There may be new information. This Instructions for Use does not take the place of talking to your child's healthcare provider about your child's medical condition or treatment.
Important information:
- Always use the oral dosing syringe provided with your child's XROMI oral solution to measure your child's prescribed dose. Ask your child’s healthcare provider or pharmacist to show you which oral dosing syringe to use and how to measure your child’s prescribed dose if you are not sure.
- Wash your hands with soap and water before and after handling XROMI oral solution and oral dosing syringes.
- Wear disposable gloves when handling XROMI oral solution and oral dosing syringes.
- Avoid contact with the oral solution. If contact with the oral solution happens on the skin, nose, or mouth, wash the area right away and thoroughly with soap and water. If contact with the oral solution happens in the eyes, flush the eyes thoroughly with water and isotonic eyewash used for that purpose for at least 15 minutes.
- If the oral solution is spilled, wipe it up right away with a damp disposable towel. Throw the damp disposable towel away in a closed container such as a plastic bag. The spill area should then be cleaned up using a detergent solution followed by clean water.
- Do not shake the bottle. Each carton of XROMI oral solution contains: • 1 bottle of XROMI oral solution • 1 bottle adapter • One 3 mL oral dosing syringe (for doses of 3 mL or less) • One 10 mL oral dosing syringe (for doses more than 3 mL)
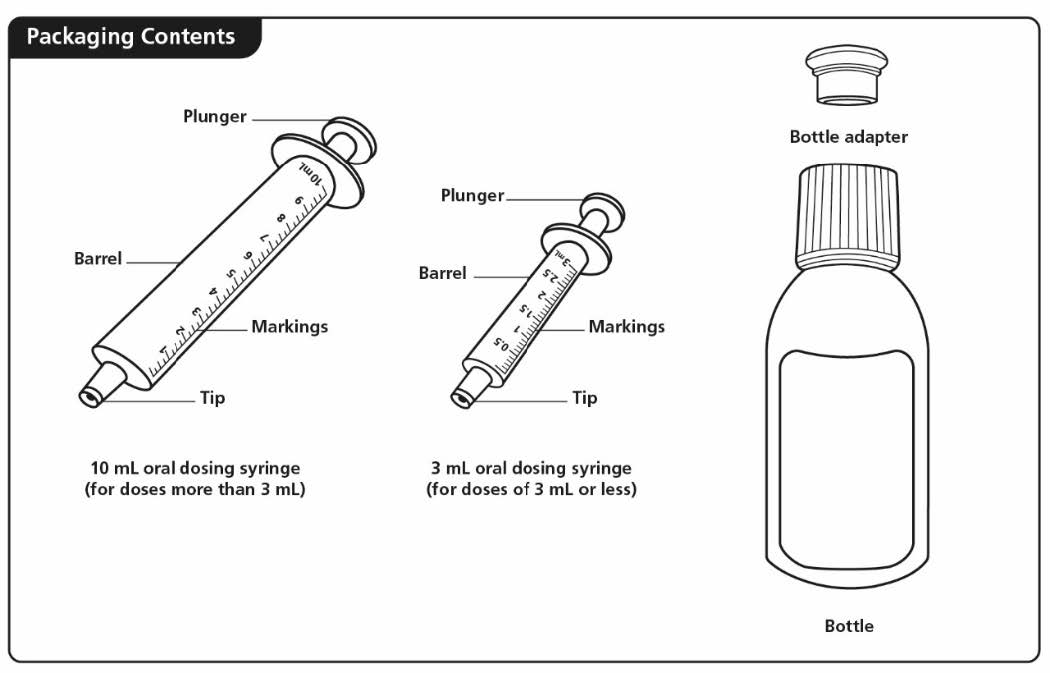
If your carton does not contain two oral dosing syringes, contact your pharmacist. You will also need disposable gloves.
| Before giving XROMI: | |
| Step 1. Wash your hands well with soap and water. | |
| Step 2. Put on disposable gloves. | |
| Step 3. Place the items from the carton on a clean flat surface. | |
| Step 4. Open the bottle by pushing down firmly on the child-resistant cap and turning it counter-clockwise (See Figure A ). Do not throw away the child-resistant cap. | 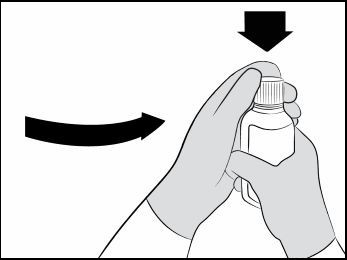 Figure A Figure A |
| Step 5. First time use only: Insert the bottle adapter. Push the ribbed end of the bottle adapter into the neck of the bottle until it is firmly in place. The bottom edge of the adapter should fully contact the top rim of the bottle (See Figure B ). Do not remove the adapter from the bottle after it is inserted. | 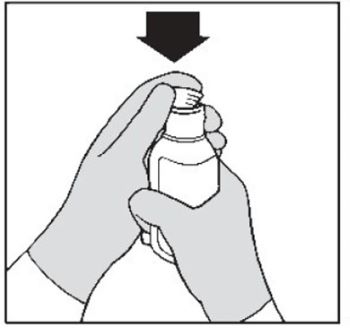 Figure B Figure B |
| Measuring your child's prescribed dose of XROMI: | |
Step 6. Choose the oral dosing syringe you need to measure your child's prescribed dose. (See Figure C ). Check the dose in mL as prescribed by your child’s healthcare provider. Choose the right oral dosing syringe for your child’s dose.
Find the marking for your child’s prescribed dose on the right oral dosing syringe. | 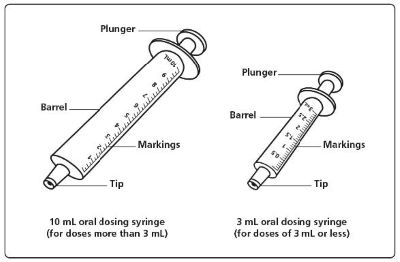 Figure C Figure C |
| Step 7. Push the plunger of the oral dosing syringe all the way down to remove any air in the oral dosing syringe (See Figure D ). | 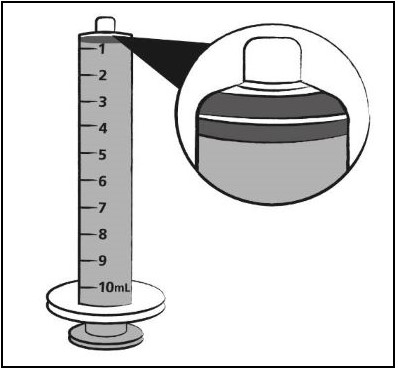 Figure D Figure D |
| Step 8. Hold the bottle upright. Insert the tip of the oral dosing syringe into the opening of the bottle adapter until it is firmly in place (See Figure E ). | 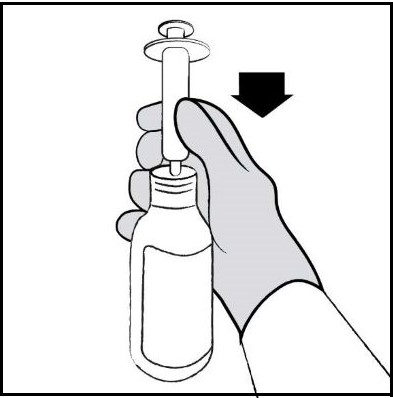 Figure E Figure E |
| Step 9. Turn the bottle upside down with the oral dosing syringe in place. Pull back slowly on the plunger of the oral dosing syringe until the top of the plunger is even with the mL mark on the oral dosing syringe that corresponds to your child’s prescribed dose. See Figure F for an example of a dose of 6 mL. Your dose may be different than the example shown. | 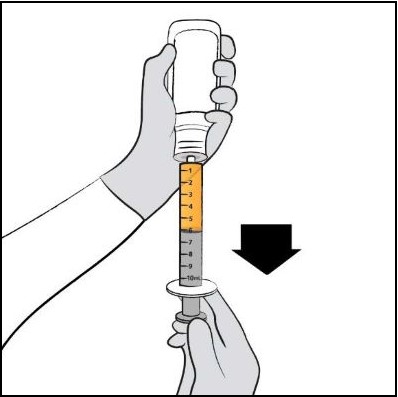 Figure F Figure F |
| Step 10. Leave the oral dosing syringe in the bottle adapter and turn the bottle upright. Place the bottle onto a flat surface. Hold the oral dosing syringe by the barrel and carefully remove it from the adapter. Do not hold the oral dosing syringe by the plunger (See Figure G ). | 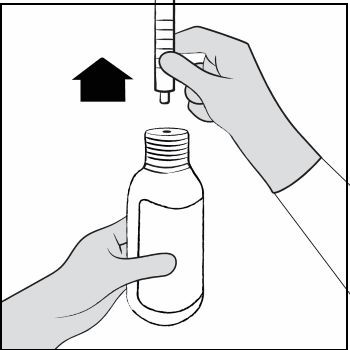 Figure G Figure G |
| Step 11. Hold the oral dosing syringe with the oral dosing syringe tip pointing up. Check that the correct dose was drawn up into the oral dosing syringe (See Figure H ). If there are large air bubbles in the oral dosing syringe (See Figure I ) or if you have drawn up the wrong dose, re-insert the oral dosing syringe tip firmly into the bottle adapter while the bottle is in an upright position. Fully push in the plunger so the oral solution flows back into the bottle. Repeat Steps 8 and 9. | 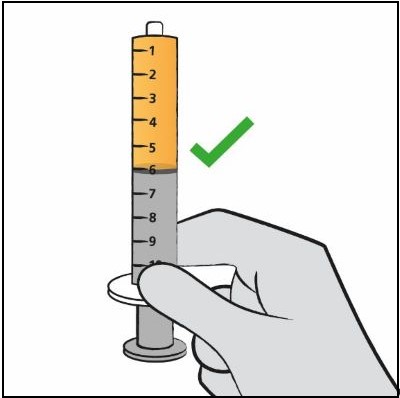 Figure H Figure H 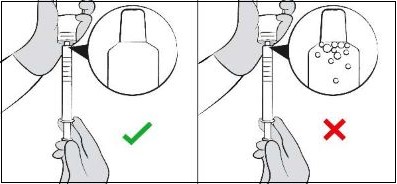 Figure I Figure I |
| Giving the prescribed dose of XROMI: | |
| Step 12. Place the tip of the oral dosing syringe in your child’s mouth and place the tip against the inside of your child’s cheek. Gently push the plunger all the way down to give all the medicine in the oral dosing syringe and then remove the oral dosing syringe from your child’s mouth (See Figure J ). Make sure the child has time to swallow the medicine. If your child’s prescribed dose is more than 10 mL, you will need to divide the dose. Follow the instructions given to you by your healthcare provider or pharmacist about how to divide the dose and repeat Steps 6 through 12. | 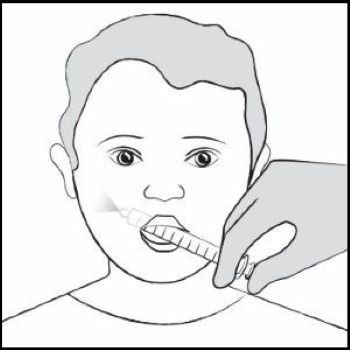 Figure J Figure J |
| Step 13. Have your child drink some water to make sure no medicine is left in your child’s mouth. | |
| Step 14. Close the bottle tightly by turning the child-resistant cap clockwise, leaving the bottle adapter in place. | |
| Step 15. Wash your hands well with soap and water. Rinse and wash the oral dosing syringes with cold or warm water and dry completely before next use. Hold the oral dosing syringe under water and move the plunger up and down several times to make sure the inside of the oral dosing syringe is clean. Let the oral dosing syringe dry completely before you use it again. Do not throw away the oral dosing syringe after use. |
Storing XROMI
- Store XROMI in a refrigerator between 35°F to 46°F (2°C to 8°C). Do not freeze.
- Store the XROMI bottle and oral dosing syringe in a clean place.
- XROMI oral solution should be used within 12 weeks after opening the bottle. Dispose of (throw away) any unused medicine and the dosing syringes after 12 weeks.
- Do not use after the expiration date on the carton and the bottle.
- Keep XROMI and all medicines out of the reach of children.
- Ask your pharmacist for instructions on how to throw away (dispose of) XROMI that is expired or no longer needed.
Manufactured by: Nova Laboratories Ltd., Leicester, LE18 4YL, United Kingdom Manufactured for: Rare Disease Therapeutics, Inc., 2550 Meridian Blvd., Suite 150, Franklin, TN 37067 Part Number: D001426/1 This “Instructions for Use” has been approved by the U.S. Food and Drug Administration Revised: 12/2024
Mechanism of Action
The precise mechanism by which hydroxyurea produces its cytotoxic and cytoreductive effects is not known. However, various studies support the hypothesis that hydroxyurea causes an immediate inhibition of DNA synthesis by acting as a ribonucleotide reductase inhibitor, without interfering with the synthesis of ribonucleic acid or of protein. The mechanisms by which XROMI produces its beneficial effects in patients with sickle cell anemia are uncertain. Known pharmacologic effects of XROMI that may contribute to its beneficial effects include increasing HbF levels in red blood cells (RBCs), decreasing neutrophils, increasing the water content of RBCs, increasing deformability of sickled cells, and altering the adhesion of RBCs to endothelium.