Get your patient on Darifenacin - Darifenacin Hydrobromide tablet, Extended Release (Darifenacin Hydrobromide)
Darifenacin - Darifenacin Hydrobromide tablet, Extended Release prescribing information
Warnings and Precautions: Central Nervous System Effects (5.5 ) 03/2012
INDICATIONS AND USAGE
Darifenacin extended-release tablets are muscarinic antagonist indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency and frequency.
DOSAGE AND ADMINISTRATION
The recommended starting dose of darifenacin extended-release tablets is 7.5 mg once daily. Based upon individual response, the dose may be increased to 15 mg once daily, as early as two weeks after starting therapy.
Darifenacin extended-release tablets should be taken once daily with water. Darifenacin extended-release tablets may be taken with or without food, and should be swallowed whole and not chewed, divided or crushed.
For patients with moderate hepatic impairment (Child-Pugh B) or when co-administered with potent CYP3A4 inhibitors (for example, ketoconazole, itraconazole, ritonavir, nelfinavir, clarithromycin and nefazadone), the daily dose of darifenacin extended-release tablets should not exceed 7.5 mg. Darifenacin extended-release tablets are not recommended for use in patients with severe hepatic impairment (Child-Pugh C) [see Warnings & Precautions (5.6) , Drug Interactions (7.1) , Use in Specific Populations (8.6) and Clinical Pharmacology (12.3) ] .
DOSAGE FORMS AND STRENGTHS
Darifenacin extended-release tablets 7.5 mg are white, round, bi-convex, film-coated, debossed with “P” on one side and “67” on the other side.
Darifenacin extended-release tablets 15 mg are light peach, round, bi-convex, film-coated, debossed with “P” on one side and “68” on the other side.
USE IN SPECIFIC POPULATIONS
- Pregnancy: Darifenacin should be used during pregnancy only if the benefit to the mother outweighs the potential risk to the fetus. (8.1 )
- Nursing Mothers: It is not known whether darifenacin is excreted into human milk and therefore caution should be exercised before darifenacin is administered to a nursing woman. (8.3 )
- Pediatric Use: The safety and effectiveness of darifenacin in pediatric patients have not been established. (8.4 )
Labor & Delivery
Pregnancy
Pregnancy Category C
There are no studies of darifenacin in pregnant women.
Darifenacin was not teratogenic in rats and rabbits at plasma exposures of free drug (via AUC) up to 59 times and 28 times, respectively (doses up to 50 and 30 mg/kg/day, respectively) the maximum recommended human dose [MRHD] of 15 mg. At approximately 59 times the MRHD in rats, there was a delay in the ossification of the sacral and caudal vertebrae which was not observed at approximately 13 times the AUC. Dystocia was observed in dams at approximately 17 times the AUC (10 mg/kg/day). Slight developmental delays were observed in pups at this dose. At five times the AUC (3 mg/kg/day), there were no effects on dams or pups. In rabbits, an exposure approximately 28 times (30 mg/kg/day) the MRHD of darifenacin was shown to increase post-implantation loss, with a no effect level at nine times (10 mg/kg/day) the AUC at the MRHD. Dilated ureter and/or kidney pelvis was also observed in offspring at this dose along with urinary bladder dilation consistent with the pharmacological action of darifenacin, with one case observed at nine times (10 mg/kg/day). No effect was observed at approximately 2.8 times (3 mg/kg/day) the AUC at the MRHD.
Because animal reproduction studies are not always predictive of human response, darifenacin should be used during pregnancy only if the benefit to the mother outweighs the potential risk to the fetus.
Nursing Mothers
Darifenacin is excreted into the milk of rats. It is not known whether darifenacin is excreted into human milk and therefore caution should be exercised before darifenacin is administered to a nursing woman.
Pediatric Use
The safety and effectiveness of darifenacin in pediatric patients have not been established.
Geriatric Use
In the fixed-dose, placebo-controlled, clinical studies, 30 percent of patients treated with darifenacin were over 65 years of age. No overall differences in safety or efficacy were observed between patients over 65 years (n = 207) and younger patients <65 years (n = 464). No dose adjustment is recommended for elderly patients [see Clinical Pharmacology (12.3) and Clinical Studies (14) ] .
Hepatic Impairment
Subjects with severe hepatic impairment (Child-Pugh C) have not been studied, therefore darifenacin is not recommended for use in these patients [see Dosage and Administration (2) and Warnings and Precautions (5.6) ] . The daily dose of darifenacin should not exceed 7.5 mg once daily for patients with moderate hepatic impairment (Child-Pugh B) [see Dosage and Administration (2) and Warnings and Precautions (5.6) ] . After adjusting for plasma protein binding, unbound darifenacin exposure was estimated to be 4.7-fold higher in subjects with moderate hepatic impairment than subjects with normal hepatic function. No dose adjustment is recommended for patients with mild hepatic impairment (Child-Pugh A).
Renal Impairment
A study of subjects with varying degrees of renal impairment (creatinine clearance between 10 and 136 mL/min) demonstrated no clear relationship between renal function and darifenacin clearance. No dose adjustment is recommended for patients with renal impairment [see Clinical Pharmacology (12.3) ] .
Gender
No dose adjustment is recommended based on gender [see Clinical Pharmacology (12.3) and Clinical Studies (14) ] .
CONTRAINDICATIONS
Darifenacin is contraindicated in patients with, or at risk for, the following conditions:
- urinary retention
- gastric retention, or
- uncontrolled narrow-angle glaucoma.
WARNINGS AND PRECAUTIONS
- Darifenacin should be administered with caution to patients with clinically significant bladder outflow obstruction because of the risk of urinary retention. (5.1 )
- Darifenacin should be administered with caution to patients with gastrointestinal obstructive disorders because of the risk of gastric retention. (5.2 )
- Darifenacin should be used with caution in patients being treated for narrow- angle glaucoma and only where the potential benefits outweigh the risks. (5.3 )
- Central Nervous System Effects: Somnolence has been reported with darifenacin. Advise patients not to drive or operate heavy machinery until they know how darifenacin affects them. (5.5 )
Risk of Urinary Retention
Darifenacin should be administered with caution to patients with clinically significant bladder outflow obstruction because of the risk of urinary retention.
Decreased Gastrointestinal Motility
Darifenacin should be administered with caution to patients with gastrointestinal obstructive disorders because of the risk of gastric retention. Darifenacin, like other anticholinergic drugs, may decrease gastrointestinal motility and should be used with caution in patients with conditions such as severe constipation, ulcerative colitis, and myasthenia gravis.
Controlled Narrow-Angle Glaucoma
Darifenacin should be used with caution in patients being treated for narrow-angle glaucoma and only where the potential benefits outweigh the risks.
Angioedema
Angioedema of the face, lips, tongue, and/or larynx have been reported with darifenacin. In some cases angioedema occurred after the first dose. Angioedema associated with upper airway swelling may be life threatening. If involvement of the tongue, hypopharynx, or larynx occurs, darifenacin should be promptly discontinued and appropriate therapy and/or measures necessary to ensure a patent airway should be promptly provided.
Central Nervous System Effects
Darifenacin is associated with anticholinergic central nervous system (CNS) effects [see Adverse Reactions (6.2) ]. A variety of CNS anticholinergic effects have been reported, including headache, confusion, hallucinations and somnolence. Patients should be monitored for signs of anticholinergic CNS effects, particularly after beginning treatment or increasing the dose. Advise patients not to drive or operate heavy machinery until they know how darifenacin affects them. If a patient experiences anticholinergic CNS effects, dose reduction or drug discontinuation should be considered.
Patients with Hepatic Impairment
The daily dose of darifenacin should not exceed 7.5 mg for patients with moderate hepatic impairment (Child-Pugh B). Darifenacin has not been studied in patients with severe hepatic impairment (Child-Pugh C) and therefore is not recommended for use in this patient population [see Dosage and Administration (2) Use in Specific Populations (8.6) and Clinical Pharmacology (12.3) ] .
ADVERSE REACTIONS
The most frequently reported adverse reactions (>3 percent) for darifenacin are: constipation, dry mouth, headache, dyspepsia, nausea, urinary tract infection, accidental injury, and flu symptoms. (6 )
To report SUSPECTED ADVERSE REACTIONS, contact Polygen Pharmaceuticals Inc. at 1-888-291-7337 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of darifenacin was evaluated in controlled clinical trials in a total of 8,830 patients, 6,001 of whom were treated with darifenacin. Of this total, 1,069 patients participated in three, 12-week, randomized, placebo-controlled, fixed-dose efficacy and safety studies (Studies 1, 2 and 3). Of this total, 337 and 334 patients received darifenacin 7.5 mg daily and 15 mg daily, respectively. In all long-term trials combined, 1,216 and 672 patients received treatment with darifenacin for at least 24 and 52 weeks, respectively.
In Studies 1, 2 and 3 combined, the serious adverse reactions to darifenacin were urinary retention and constipation.
In Studies 1, 2 and 3 combined, dry mouth leading to study discontinuation occurred in 0 percent, 0.9 percent, and 0 percent of patients treated with darifenacin 7.5 mg daily, darifenacin 15 mg daily and placebo, respectively.
Constipation leading to study discontinuation occurred in 0.6 percent, 1.2 percent, and 0.3 percent of patients treated with darifenacin 7.5 mg daily, darifenacin 15 mg daily and placebo, respectively.
Table 1 lists the rates of identified adverse reactions, derived from all reported adverse events in 2 percent or more of patients treated with 7.5 mg or 15 mg darifenacin, and greater than placebo in Studies 1, 2 and 3. In these studies, the most frequently reported adverse reactions were dry mouth and constipation. The majority of the adverse reactions were mild or moderate in severity and most occurred during the first two weeks of treatment.
| Body System | Adverse Reaction | Percentage of Subjects | ||
| Darifenacin 7.5 mg N = 337 | Darifenacin 15 mg N = 334 | Placebo N = 388 | ||
| Digestive | Dry Mouth | 20.2 | 35.3 | 8.2 |
| Constipation | 14.8 | 21.3 | 6.2 | |
| Dyspepsia | 2.7 | 8.4 | 2.6 | |
| Abdominal Pain | 2.4 | 3.9 | 0.5 | |
| Nausea | 2.7 | 1.5 | 1.5 | |
| Diarrhea | 2.1 | 0.9 | 1.8 | |
| Urogenital | Urinary Tract Infection | 4.7 | 4.5 | 2.6 |
| Nervous | Dizziness | 0.9 | 2.1 | 1.3 |
| Body as whole | Asthenia | 1.5 | 2.7 | 1.3 |
| Eye | Dry Eyes | 1.5 | 2.1 | 0.5 |
Other adverse reactions reported by 1 percent to 2 percent of darifenacin-treated patients include: abnormal vision, accidental injury, back pain, dry skin, flu syndrome, hypertension, vomiting, peripheral edema, weight gain, arthralgia, bronchitis, pharyngitis, rhinitis, sinusitis, rash, pruritus, urinary tract disorder and vaginitis.
Study 4 was a randomized, 12-week, placebo-controlled, dose-titration regimen study in which darifenacin was administered in accordance with dosing recommendations [see Dosage and Administration (2) ] . All patients initially received placebo or darifenacin 7.5 mg daily, and after two weeks, patients and physicians were allowed to adjust upward to darifenacin 15 mg if needed. In this study, the most commonly reported adverse reactions were also constipation and dry mouth. Table 2 lists the identified adverse reactions, derived from all adverse events reported in >3 percent of patients treated with darifenacin and greater than placebo.
| Adverse Reaction | Darifenacin 7.5 mg/15 mg N = 268 | Placebo N = 127 |
| Constipation | 56 (20.9 percent) | 10 (7.9 percent) |
| Dry Mouth | 50 (18.7 percent) | 11 (8.7 percent) |
| Headache | 18 (6.7 percent) | 7 (5.5 percent) |
| Dyspepsia | 12 (4.5 percent) | 2 (1.6 percent) |
| Nausea | 11 (4.1 percent) | 2 (1.6 percent) |
| Urinary Tract Infection | 10 (3.7 percent) | 4 (3.1 percent) |
| Accidental Injury | 8 (3.0 percent) | 3 (2.4 percent) |
| Flu Syndrome | 8 (3.0 percent) | 3 (2.4 percent) |
Post Marketing Experience
The following adverse reactions have been reported during post approval use of darifenacin extended-release tablets (darifenacin). Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate frequency or establish a causal relationship to drug exposure.
Dermatologic: erythema multiforme, interstitial granuloma annulare
General: hypersensitivity reactions, including angioedema with airway obstruction and anaphylactic reaction
Central Nervous: confusion, hallucinations and somnolence
Cardiovascular: palpitations and syncope
DRUG INTERACTIONS
- Caution should be taken when darifenacin is used concomitantly with medications that are predominantly metabolized by CYP2D6 and which have a narrow therapeutic window, such as flecainide, thioridazine and tricyclic antidepressants. (7.2 )
- The concomitant use of darifenacin with other anticholinergic agents may increase the frequency and/or severity of dry mouth, constipation, blurred vision and other anticholinergic pharmacological effects. Anticholinergic agents may potentially alter the absorption of some concomitantly administered drugs due to effects of gastrointestinal motility. (7.3 )
CYP3A4 Inhibitors
The systemic exposure of darifenacin from darifenacin extended-release tablets is increased in the presence of CYP3A4 inhibitors. The daily dose of darifenacin should not exceed 7.5 mg when co-administered with potent CYP3A4 inhibitors (for example, ketoconazole, itraconazole, ritonavir, nelfinavir, clarithromycin and nefazadone). No dosing adjustments are recommended in the presence of moderate CYP3A4 inhibitors (for example, erythromycin, fluconazole, diltiazem and verapamil) [see Dosage and Administration (2) and Clinical Pharmacology (12.3) ] .
CYP2D6 Inhibitors
No dosing adjustments are recommended in the presence of CYP2D6 inhibitors (for example, paroxetine, fluoxetine, quinidine and duloxetine) [see Clinical Pharmacology (12.3) ] .
CYP2D6 Substrates
Caution should be taken when darifenacin is used concomitantly with medications that are predominantly metabolized by CYP2D6 and which have a narrow therapeutic window (for example, flecainide, thioridazine and tricyclic antidepressants) [see Clinical Pharmacology (12.3) ] .
CYP3A4 Substrates
Darifenacin (30 mg daily) did not have a significant impact on midazolam (7.5 mg) pharmacokinetics [see Clinical Pharmacology (12.3) ] .
Combination oral contraceptives
Darifenacin (10 mg three times daily) had no effect on the pharmacokinetics of the combination oral contraceptives containing levonorgestrel and ethinyl estradiol [see Clinical Pharmacology (12.3) ] .
Warfarin
Darifenacin had no significant effect on prothrombin time when a single dose of warfarin 30 mg was co-administered with darifenacin (30 mg daily) at steady-state. Standard therapeutic prothrombin time monitoring for warfarin should be continued.
Digoxin
Darifenacin (30 mg daily) did not have a clinically relevant effect on the pharmacokinetics of digoxin (0.25 mg) at steady-state. Routine therapeutic drug monitoring for digoxin should be continued [see Clinical Pharmacology (12.3) ] .
Other Anticholinergic Agents
The concomitant use of darifenacin with other anticholinergic agents may increase the frequency and/or severity of dry mouth, constipation, blurred vision and other anticholinergic pharmacological effects. Anticholinergic agents may potentially alter the absorption of some concomitantly administered drugs due to effects on gastrointestinal motility.
DESCRIPTION
Darifenacin is an extended-release tablet for oral administration which contains 7.5 mg or 15 mg darifenacin as its hydrobromide salt. The active moiety, darifenacin, is a potent muscarinic receptor antagonist.
Chemically, darifenacin hydrobromide is (S) -2-{1-[2-(2,3-dihydrobenzofuran-5-yl)ethyl]-3-pyrrolidinyl}-2,2-diphenylacetamide hydrobromide. The molecular formula of darifenacin hydrobromide is C 28 H 30 N 2 O 2 •HBr.
The structural formula is:
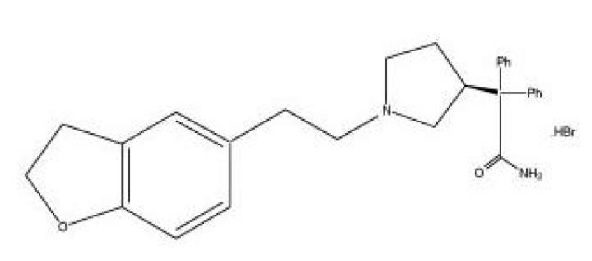
Darifenacin hydrobromide is a white to off-white, crystalline powder, with a molecular weight of 507.5.
Darifenacin is a once-a-day extended-release tablet and contains the following inactive ingredients: dibasic calcium phosphate anhydrous, hypromellose, magnesium stearate, polyethylene glycol, talc, titanium dioxide. The 15 mg tablet also contains iron oxide red and iron oxide yellow.
CLINICAL PHARMACOLOGY
Mechanism of Action
Darifenacin is a competitive muscarinic receptor antagonist. Muscarinic receptors play an important role in several major cholinergically mediated functions, including contractions of the urinary bladder smooth muscle and stimulation of salivary secretion.
In vitro studies using human recombinant muscarinic receptor subtypes show that darifenacin has greater affinity for the M 3 receptor than for the other known muscarinic receptors (9- and 12-fold greater affinity for M 3 compared to M 1 and M 5 , respectively, and 59-fold greater affinity for M 3 compared to both M 2 and M 4 ). M 3 receptors are involved in contraction of human bladder and gastrointestinal smooth muscle, saliva production, and iris sphincter function. Adverse drug effects such as dry mouth, constipation and abnormal vision may be mediated through effects on M 3 receptors in these organs.
Pharmacodynamics
In three cystometric studies performed in patients with involuntary detrusor contractions, increased bladder capacity was demonstrated by an increased volume threshold for unstable contractions and diminished frequency of unstable detrusor contractions after darifenacin treatment. These findings are consistent with an antimuscarinic action on the urinary bladder.
Electrophysiology
The effect of six-day treatment of 15 mg and 75 mg darifenacin on QT/QTc interval was evaluated in a multiple-dose, double-blind, randomized, placebo- and active-controlled (moxifloxacin 400 mg) parallel-arm design study in 179 healthy adults (44 percent male, 56 percent female) aged 18 to 65. Subjects included 18 percent poor metabolizer (PMs) and 82 percent extensive metabolizer (EMs). The QT interval was measured over a 24-hour period both pre-dosing and at steady-state. The 75 mg darifenacin dose was chosen because this achieves exposure similar to that observed in CYP2D6 poor metabolizers administered the highest recommended dose (15 mg) of darifenacin in the presence of a potent CYP3A4 inhibitor. At the doses studied, darifenacin did not result in QT/QTc interval prolongation at any time during the steady-state, while moxifloxacin treatment resulted in a mean increase from baseline QTcF of about 7.0 msec when compared to placebo. In this study, darifenacin 15 mg and 75 mg doses demonstrated a mean heart rate change of 3.1 and 1.3 bpm, respectively, when compared to placebo. However, in the clinical efficacy and safety studies, the change in median HR following treatment with darifenacin was no different from placebo.
Pharmacokinetics
Absorption
After oral administration of darifenacin to healthy volunteers, peak plasma concentrations of darifenacin are reached approximately seven hours after multiple dosing and steady-state plasma concentrations are achieved by the sixth day of dosing. The mean (SD) steady-state time course of darifenacin 7.5 mg and 15 mg extended-release tablets is depicted in Figure 1.
Figure 1: Mean (SD) Steady-State Darifenacin Plasma Concentration-Time Profiles for Darifenacin 7.5 mg and 15 mg in Healthy Volunteers Including Both CYP2D6 EMs and PMs •
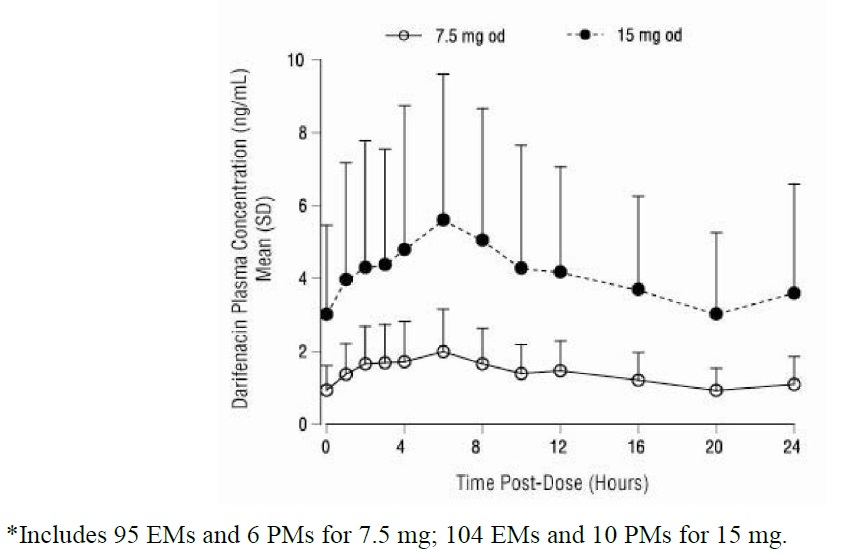
A summary of mean (standard deviation, SD) steady-state pharmacokinetic parameters of darifenacin 7.5 mg and 15 mg extended-release tablets in EMs and PMs of CYP2D6 is provided in Table 3.
| Darifenacin 7.5 mg (N = 68 EM, 5 PM) | Darifenacin 15 mg (N = 102 EM, 17 PM) | |||||||||
| AUC 2 4 ( ng • h / mL ) | C m a x ( ng / mL ) | C a v g ( ng / mL ) | T m a x ( h ) | t 1 / 2 ( h ) | AUC 2 4 ( ng • h / mL ) | C m a x ( ng / mL ) | C a v g ( ng / mL ) | T m a x ( h ) | t 1 / 2 ( h ) | |
| EM | 29.24 (15.47) | 2.01 (1.04) | 1.22 (0.64) | 6.49 (4.19) | 12.43 (5.64) a | 88.90 (67.87) | 5.76 (4.24) | 3.70 (2.83) | 7.61 (5.06) | 12.05 (12.37) b |
| PM | 67.56 (13.13) | 4.27 (0.98) | 2.81 (0.55) | 5.20 (1.79) | 19.95 c - | 157.71 (77.08) | 9.99 (5.09) | 6.58 (3.22) | 6.71 (3.58) | 7.40 d - |
| a N = 25; b N = 8; c N = 2; d N = 1; AUC 2 4 = Area under the plasma concentration versus time curve for 24h; | ||||||||||
| C m a x = Maximum observed plasma concentration; C a v g = Average plasma concentration at steady-state; | ||||||||||
| T m a x = Time of occurrence of C m a x ; t 1 / 2 = Terminal elimination half-life. Regarding EM and PM [see CLINICAL PHARMACOLOGY, Pharmacokinetics, Variability in Metabolism (12.3)]. | ||||||||||
The mean oral bioavailability of darifenacin in EMs at steady-state is estimated to be 15 percent and 19 percent for mg and 15 mg tablets, respectively.
Effect of Food
Following single dose administration of darifenacin with food, the AUC of darifenacin was not affected, while the C max was increased by 22 percent and T max was shortened by 3.3 hours. There is no effect of food on multiple-dose pharmacokinetics from darifenacin.
Distribution
Darifenacin is approximately 98 percent bound to plasma proteins (primarily to alpha-1-acid-glycoprotein). The steady-state volume of distribution (V ss ) is estimated to be 163 L.
Metabolism
Darifenacin is extensively metabolized by the liver following oral dosing.
Metabolism is mediated by cytochrome P450 enzymes CYP2D6 and CYP3A4. The three main metabolic routes are as follows:
(i) monohydroxylation in the dihydrobenzofuran ring;
(ii) dihydrobenzofuran ring opening;
(iii) N-dealkylation of the pyrrolidine nitrogen.
The initial products of the hydroxylation and N-dealkylation pathways are the major circulating metabolites but they are unlikely to contribute significantly to the overall clinical effect of darifenacin.
Variability in Metabolism
A subset of individuals (approximately 7 percent Caucasians and 2 percent African Americans) are poor metabolizers (PMs) of CYP2D6 metabolized drugs. Individuals with normal CYP2D6 activity are referred to as extensive metabolizers (EMs). The metabolism of darifenacin in PMs will be principally mediated via CYP3A4. The darifenacin ratios (PM versus EM) for C max and AUC following darifenacin 15 mg once daily at steady-state were 1.9 and 1.7, respectively.
Excretion
Following administration of an oral dose of 14 C-darifenacin solution to healthy volunteers, approximately 60 percent of the radioactivity was recovered in the urine and 40 percent in the feces. Only a small percentage of the excreted dose was unchanged darifenacin (3 percent). Estimated darifenacin clearance is 40 L/h for EMs and 32 L/h for PMs. The elimination half-life of darifenacin following chronic dosing is approximately 13 to 19 hours.
Drug-Drug Interactions
Effects of Other Drugs on Darifenacin
Darifenacin metabolism is primarily mediated by the cytochrome P450 enzymes CYP2D6 and CYP3A4. Therefore, inducers of CYP3A4 or inhibitors of either of these enzymes may alter darifenacin pharmacokinetics [see Drug Interactions (7) ] .
CYP3A4 Inhibitors: In a drug interaction study, when a 7.5 mg once daily dose of darifenacin was given to steady-state and co-administered with the potent CYP3A4 inhibitor ketoconazole 400 mg, mean darifenacin Cmax increased to 11.2 ng/mL for EMs (n = 10) and 55.4 ng/mL for one PM subject (n = 1). Mean AUC increased to 143 and 939 ng • h/mL for EMs and for one PM subject, respectively. When a 15 mg daily dose of darifenacin was given with ketoconazole, mean darifenacin Cmax increased to 67.6 ng/mL and 58.9 ng/mL for EMs (n = 3) and one PM subject (n = 1), respectively. Mean AUC increased to 1,110 and 931 ng • h/mL for EMs and for one PM subject, respectively [see Dosage and Administration (2) and Drug Interactions (7.1) ] .
The mean C max and AUC of darifenacin following 30 mg once daily dosing at steady-state were 128 percent and 95 percent higher, respectively, in the presence of a moderate CYP3A4 inhibitor, erythromycin. Co-administration of fluconazole, a moderate CYP3A4 inhibitor and darifenacin 30 mg once daily at steady-state increased darifenacin C max and AUC by 88 percent and 84 percent, respectively [see Drug Interactions (7.1) ] .
The mean C max and AUC of darifenacin following 30 mg once daily at steady-state were 42 percent and 34 percent higher, respectively, in the presence of cimetidine, a mixed CYP P450 enzyme inhibitor.
CYP2D6 Inhibitors: Darifenacin exposure following 30 mg once daily at steady-state was 33 percent higher in the presence of the potent CYP2D6 inhibitor paroxetine 20 mg [see Drug Interactions (7.2) ] .
Effects of Darifenacin on Other Drugs
In Vitro Studies: Based on in vitro human microsomal studies, darifenacin is not expected to inhibit CYP1A2 or CYP2C9 at clinically relevant concentrations.
In Vivo Studies: The potential for clinical doses of darifenacin to act as inhibitors of CYP2D6 or CYP3A4 substrates was investigated in specific drug interaction studies.
CYP2D6 Substrates: The mean C max and AUC of imipramine, a CYP2D6 substrate, were increased by 57 percent and 70 percent, respectively, in the presence of steady-state darifenacin 30 mg once daily. The mean C max and AUC of desipramine, the active metabolite of imipramine, were increased by 260 percent [see Drug Interactions (7.3) ] .
CYP3A4 Substrates: Darifenacin (30 mg daily) co-administered with a single oral dose of midazolam 7.5 mg resulted in a 17 percent increase in midazolam exposure.
Combination Oral Contraceptives: Darifenacin (10 mg three times daily) had no effect on the pharmacokinetics of a combination oral contraceptive containing levonorgestrel (0.15 mg) and ethinyl estradiol (0.03 mg).
Warfarin: Darifenacin had no significant effect on prothrombin time when a single dose of warfarin 30 mg was co-administered with darifenacin (30 mg daily) at steady-state [see Drug Interactions (7.6) ] .
Digoxin: Darifenacin (30 mg daily) co-administered with digoxin (0.25 mg) at steady-state resulted in a 16 percent increase in digoxin exposure [see Drug Interactions (7.7) ] .
Pharmacokinetics in Special Populations
Age: A population pharmacokinetic analysis of patient data indicated a trend for clearance of darifenacin to decrease with age (6 percent per decade relative to a median age of 44). Following administration of darifenacin 15 mg once daily, darifenacin exposure at steady-state was approximately 12 percent to 19 percent higher in volunteers between 45 and 65 years of age compared to younger volunteers aged 18 to 44 years [see Use in Specific Populations (8.5) ].
Pediatric: The pharmacokinetics of darifenacin has not been studied in the pediatric population [see Use in Specific Populations (8.4) ] .
Gender: PK parameters were calculated for 22 male and 25 female healthy volunteers. Darifenacin C max and AUC at steady-state were approximately 57 percent to 79 percent and 61 percent to 73 percent higher in females than in males, respectively [see Use in Specific Populations (8.8) ] .
Renal Impairment: A study of subjects with varying degrees of renal impairment (creatinine clearance between 10 and 136 mL/min) given darifenacin 15 mg once daily to steady-state demonstrated no clear relationship between renal function and darifenacin clearance [see Use in Specific Populations (8.7) ] .
Hepatic Impairment: Darifenacin pharmacokinetics were investigated in subjects with mild (Child-Pugh A) or moderate (Child-Pugh B) impairment of hepatic function given darifenacin 15 mg once daily to steady-state. Mild hepatic impairment had no effect on the pharmacokinetics of darifenacin. However, protein binding of darifenacin was affected by moderate hepatic impairment. After adjusting for plasma protein binding, unbound darifenacin exposure was estimated to be 4.7-fold higher in subjects with moderate hepatic impairment than subjects with normal hepatic function. Subjects with severe hepatic impairment (Child-Pugh C) have not been studied [see Dosage and Administration (2) , Warning and Precautions (5.5) and Use in Specific Population (8.6) ] .
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with darifenacin were conducted in mice and rats. No evidence of drug-related carcinogenicity was revealed in a 24-month study in mice at dietary doses up to 100 mg/kg/day or approximately 32 times the estimated free plasma AUC reached at the maximum recommended human dose (the AUC at the MRHD) of 15 mg and in a 24-month study in rats at doses up to 15 mg/kg/day or up to approximately 12 times the AUC at the MRHD in female rats and approximately eight times the AUC at the MRHD in male rats.
Darifenacin was not genotoxic in the bacterial mutation assay (Ames test), the Chinese hamster ovary assay, the human lymphocyte assay, or the in vivo mouse bone marrow cytogenetics assay.
There was no evidence for effects on fertility in male or female rats treated at oral doses up to approximately 78 times (50 mg/kg/day) the AUC at the MRHD.
CLINICAL STUDIES
Darifenacin extended-release tablets were evaluated for the treatment of patients with overactive bladder with symptoms of urgency, urge urinary incontinence, and increased urinary frequency in three randomized, fixed-dose, placebo-controlled, multicenter, double-blind, 12-week studies (Studies 1, 2 and 3) and one randomized, double-blind, placebo-controlled, multicenter, dose-titration study (Study 4). For study eligibility in all four studies, patients with symptoms of overactive bladder for at least six months were required to demonstrate at least eight micturitions and at least one episode of urinary urgency per day, and at least five episodes of urge urinary incontinence per week. The majority of patients were white (94 percent) and female (84 percent), with a mean age of 58 years, range 19 to 93 years. Thirty-three percent of patients were ≥65 years of age. These characteristics were well balanced across treatment groups. The study population was inclusive of both naïve patients who had not received prior pharmacotherapy for overactive bladder (60 percent) and those who had (40 percent).
Table 4 shows the efficacy data collected from 7- or 14-day voiding diaries in the three fixed-dose placebo-controlled studies of 1,059 patients treated with placebo, 7.5 mg or 15 mg once daily darifenacin for 12 weeks. A significant decrease in the primary endpoint, change from baseline in average weekly urge urinary incontinence episodes was observed in all three studies. Data is also shown for two secondary endpoints, change from baseline in the average number of micturitions per day (urinary frequency) and change from baseline in the average volume voided per micturition. 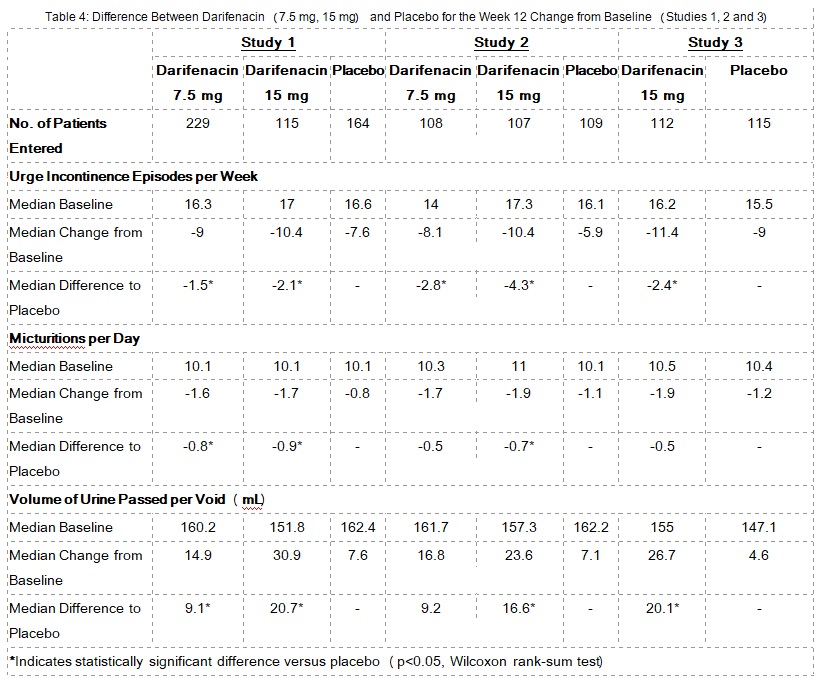
Table 5 shows the efficacy data from the dose-titration study in 395 patients who initially received 7.5 mg darifenacin or placebo daily with the option to increase to 15 mg darifenacin or placebo daily after two weeks. 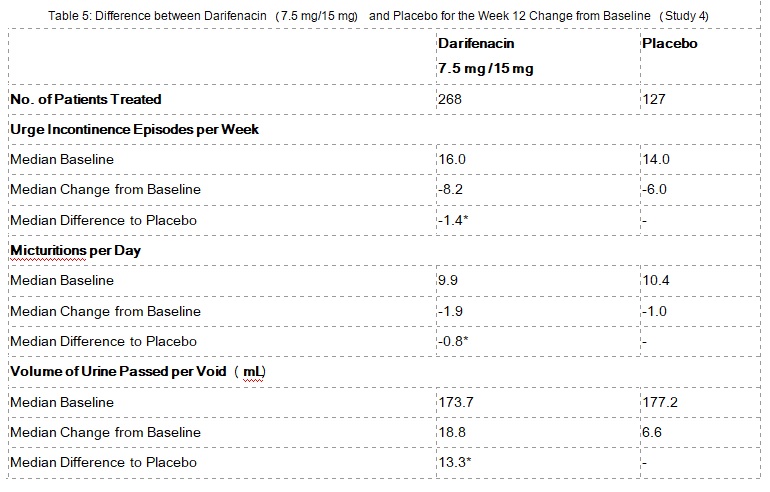
•Indicates statistically significant difference versus placebo (p<0.05, Wilcoxon rank-sum test)
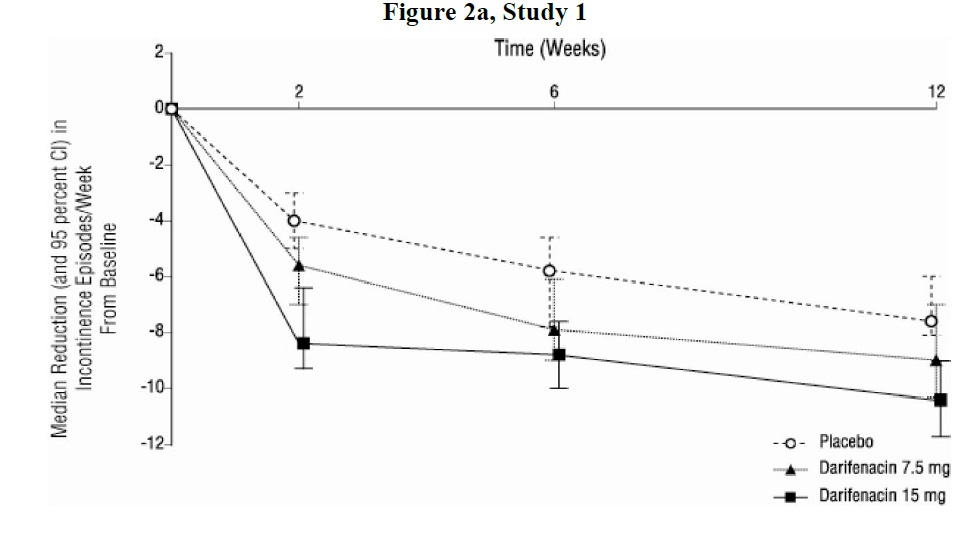
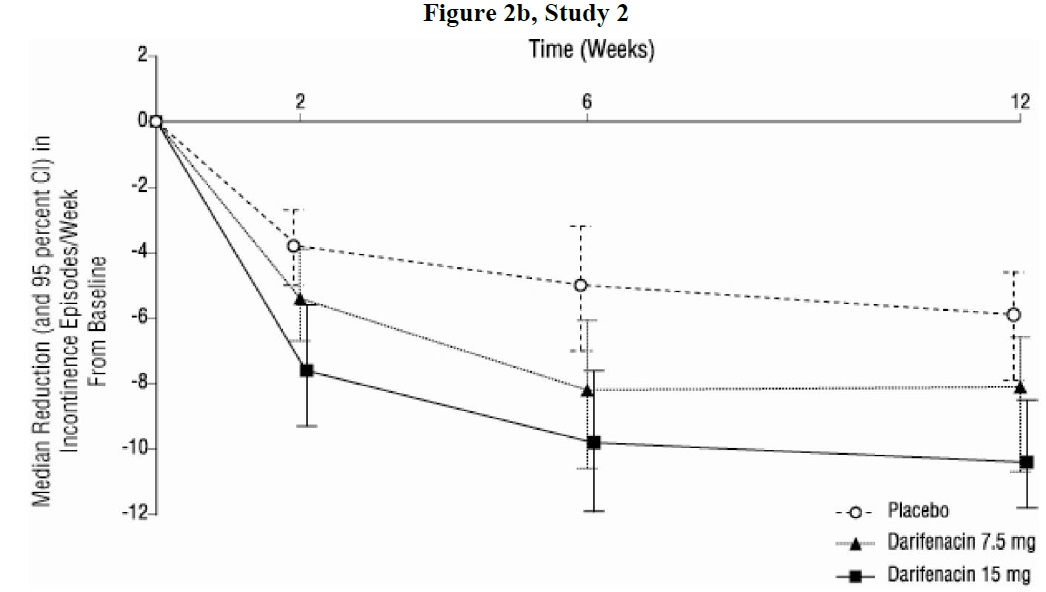
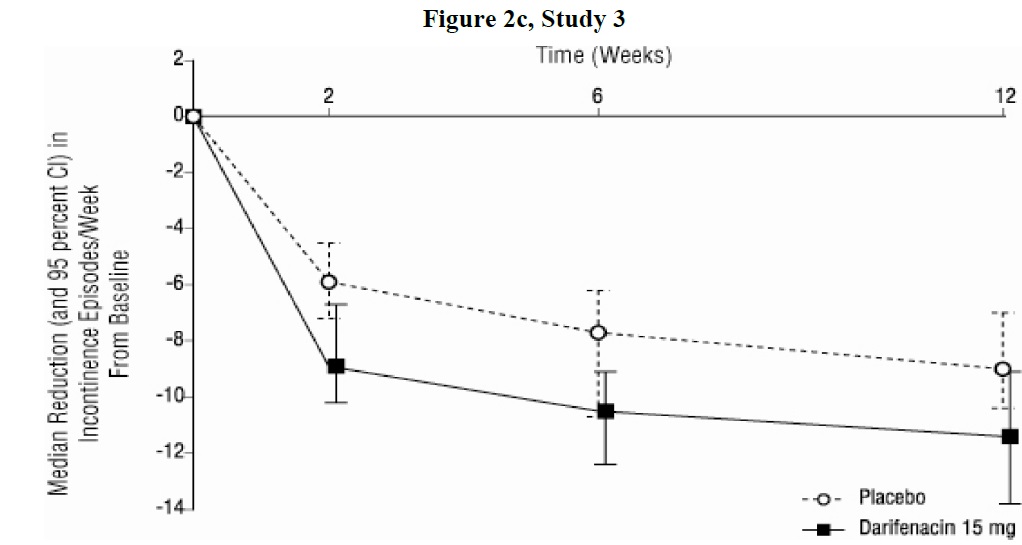
As seen in Figures 2 a, b and c, reductions in the number of urge incontinence episodes per week were observed within the first two weeks in patients treated with darifenacin 7.5 mg and 15 mg once daily compared to placebo. Further, these effects were sustained throughout the 12-week treatment period.
Figures 2a, 2b, 2c. Median Change from Baseline at Weeks 2, 6, 12 for Number of Urge Incontinence Episodes per Week (Studies 1, 2 and 3)
HOW SUPPLIED/STORAGE AND HANDLING
Darifenacin extended-release tablets ,7.5 mg are white, round, bi-convex, film-coated, debossed with “P” on one side and “67” on the other side.
Bottle of 90 with child-resistant closure.................................................................. NDC 52605-067-19
Bottle of 1000 ......................................................................................................... NDC 52605-067-10
Darifenacin extended-release tablets, 15 mg are light peach, round, bi-convex, film-coated, debossed with “P” on one side and “68” on the other side.
Bottle of 30 with child-resistant closure.................................................................. NDC 52605-068-13
Bottle of 90 with child-resistant closure.................................................................. NDC 52605-068-19
Bottle of 1000 ......................................................................................................... NDC 52605-068-10
Storage
Store at 25° C (77° F) [See USP Controlled Room Temperature]. Protect from light.
Keep this and all drugs out of the reach of children.
Mechanism of Action
Darifenacin is a competitive muscarinic receptor antagonist. Muscarinic receptors play an important role in several major cholinergically mediated functions, including contractions of the urinary bladder smooth muscle and stimulation of salivary secretion.
In vitro studies using human recombinant muscarinic receptor subtypes show that darifenacin has greater affinity for the M 3 receptor than for the other known muscarinic receptors (9- and 12-fold greater affinity for M 3 compared to M 1 and M 5 , respectively, and 59-fold greater affinity for M 3 compared to both M 2 and M 4 ). M 3 receptors are involved in contraction of human bladder and gastrointestinal smooth muscle, saliva production, and iris sphincter function. Adverse drug effects such as dry mouth, constipation and abnormal vision may be mediated through effects on M 3 receptors in these organs.