Get your patient on Emrelis (Telisotuzumab Vedotin)
Patient education
Administration guides
Patient education materials
Patient support program
Dosing resources
Clinical information
Insurance resources
Prior authorization & coverage support
Reimbursement information
Other resources
Dosage & administration

Emrelis prescribing information
1 INDICATIONS AND USAGE
EMRELIS is indicated for the treatment of adult patients with locally advanced or metastatic, non-squamous non-small cell lung cancer (NSCLC) with high c-Met protein overexpression [≥50% of tumor cells with strong (3+) staining], as determined by an FDA-approved test [see Dosage and Administration (2.1 )] , who have received a prior systemic therapy .
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR) [see Clinical Studies (14 )] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
2 DOSAGE AND ADMINISTRATION
For intravenous infusion only. (2.5 )
2.1 Patient Selection
Select patients for treatment with EMRELIS based on the presence of high c-Met protein overexpression [≥50% of tumor cells with strong (3+) staining] in patients with non-squamous NSCLC [see Indications and Usage (1 ) and Clinical Studies (14 )] .
Information on FDA-approved tests for the detection of high c-Met protein overexpression is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of EMRELIS is 1.9 mg/kg (up to a maximum of 190 mg for patients greater than or equal to 100 kg) administered as an intravenous infusion over 30 minutes every 2 weeks until disease progression or unacceptable toxicity.
2.3 Dosage Modifications for Adverse Reactions
The recommended dose reductions for adverse reactions are provided in Table 1.
| Dose Reduction | Recommended Dosage |
| First | 1.6 mg/kg every 2 weeks |
| Second | 1.3 mg/kg every 2 weeks |
| Third | 1 mg/kg every 2 weeks |
| Permanently discontinue EMRELIS in patients who are unable to tolerate 1 mg/kg. | |
The recommended dosage modifications of EMRELIS for adverse reactions are provided in Table 2.
| Adverse Reaction | Severity a | Dosage Modification |
| Peripheral Neuropathy [see Warnings and Precautions (5.1 )] | Grade 2 or 3 |
|
| Grade 4 | Permanently discontinue EMRELIS. | |
| Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.2 )] | Grade 1 |
|
| Grade ≥2 | Permanently discontinue EMRELIS. | |
| Keratitis [see Warnings and Precautions (5.3 )] | Grade 2 |
|
| Grade 3 or 4 |
| |
| Infusion-Related Reactions (IRR) [see Warnings and Precautions (5.4 )] | Grade 1-3 |
|
| Grade 4 |
| |
| Peripheral Edema [see Adverse Reactions (6.1 )] | Grade ≥2 | First occurrence
|
| Other Adverse Reactions [see Adverse Reactions (6.1 )] | Grade 3 | First occurrence
|
| Grade 4 |
| |
| a Adverse reactions were graded using National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03 | ||
2.000000000000000e+00 4 Recommended Premedications for Patients Who Experience Infusion-Related Reactions
Table 3 contains the recommended premedications for patients who experience infusion-related reactions to EMRELIS, for subsequent infusions.
| Medication Class | E xamples or equivalent | Dose | Route of Administration | Dosing Window Prior to EMRELIS Administration |
| H1 Antihistamine | Diphenhydramine | 25 to 50 mg | Intravenously or orally | Administer 30-60 minutes prior to each infusion |
| H2 Antihistamine | Famotidine | 20 mg | Intravenously or orally | Administer 30-60 minutes prior to each infusion |
| Antipyretic | Acetaminophen | 650 to 1,000 mg | Intravenously or orally | Administer 30-60 minutes prior to each infusion |
| Glucocorticoid | Methylprednisolone | 125 mg or equivalent | Intravenously | Administer 30-60 minutes prior to each infusion |
2.000000000000000e+00 5 Preparation and Administration
EMRELIS contains a hazardous component. Follow applicable special handling and disposal procedures in accordance with local requirements. 1
Reconstitute and further dilute EMRELIS prior to intravenous infusion.
Reconstitution of Lyophilized EMRELIS
Before reconstitution, allow the vial to reach room temperature after removal of the vial from storage condition.
- Calculate the recommended dose based on the patient’s weight to determine the number of vials needed. For patients weighing greater than or equal to 100 kg, use 190 mg dose [see Dosage and Administration (2.2 )] . More than one vial may be needed to achieve the calculated dose.
- Using a sterile syringe, slowly inject Sterile Water for Injection, using the volume provided in Table 4, into the EMRELIS vial containing the lyophilized powder, which has a whole or fragmented cake-like appearance. The reconstituted solution has a concentration of 20 mg/mL EMRELIS.
| Dose Vial | Volume of Sterile Water for Injection required for reconstitution |
| 20 mg vial | 1.1 mL |
| 100 mg vial | 5.2 mL |
- Swirl the vial gently until completely dissolved. Do not shake.
- Inspect the reconstituted solution for particulate matter and discoloration. The solution should appear clear to slightly opalescent and colorless to slightly yellow. Discard the vial if the reconstituted solution is discolored, is cloudy, or contains visible particulates.
- Use reconstituted EMRELIS immediately. If not used immediately, store the reconstituted EMRELIS vials in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours from the time of reconstitution. Do not freeze.
- Each vial of EMRELIS is intended for one-time use only. Discard any unused drug remaining in the vial.
Dilution in Infusion Bag
- Calculate the required dose volume (mL) of reconstituted EMRELIS solution based on the prescribed dose.
- Withdraw the calculated dose volume (mL) of reconstituted solution from the EMRELIS vial using a sterile syringe. Discard any unused portion remaining in the vial(s).
- Inject the calculated amount of reconstituted solution into 0.9% Sodium Chloride Injection infusion bag so that the final EMRELIS concentration is between 1 mg/mL and 10 mg/mL. Use only 0.9% Sodium Chloride Injection.
- Gently invert the infusion bag to thoroughly mix the solution. Do not shake.
- After preparing the dose for infusion, visually inspect the bag content for particulates and discard if present.
- If not used immediately, the diluted solution can be stored in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours and an additional 4 hours at room temperature at 9°C to 30°C (48°F to 86°F) until the end of administration. Do not freeze.
Method of Administration
If the prepared infusion solution was stored refrigerated at 2°C to 8°C (36°F to 46°F), allow the solution to reach room temperature prior to administration.
- Administer by intravenous infusion over 30 minutes using a dedicated infusion line with a 0.20 or 0.22 micron in-line filter made of polyether sulfone (PES), polyvinylidene fluoride (PVDF), or Polyamide (PA).
- Do not mix EMRELIS with other drugs or administer other drugs through the same intravenous line.
3 DOSAGE FORMS AND STRENGTHS
For injection: 20 mg or 100 mg of telisotuzumab vedotin-tllv as a white to off-white, lyophilized powder in a single-dose vial for reconstitution and further dilution.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on the mechanism of action and findings in animals, EMRELIS can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1 )] . There are no available human data on EMRELIS use in pregnant women to inform a drug-associated risk. In an animal reproduction study, administration of the small molecule component of EMRELIS, MMAE, to pregnant rats during organogenesis resulted in embryo-fetal mortality and structural abnormalities at exposures similar to the clinical exposure at the recommended dose [see Data ] . Advise patients of the potential risks to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
No embryo-fetal development studies in animals have been performed with telisotuzumab vedotin-tllv. In an embryo-fetal development study in pregnant rats, administration of two intravenous doses of MMAE, the small molecule component of EMRELIS, on gestational days 6 and 13 caused embryo-fetal mortality and structural abnormalities, including protruding tongue, agnathia, malrotated limbs, and gastroschisis compared to controls at a dose of 0.2 mg/kg (approximately 2 times the human area under the curve [AUC] at the recommended dose).
8.2 Lactation
Risk Summary
There are no data on the presence of telisotuzumab vedotin-tllv or MMAE in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise lactating women not to breastfeed during treatment with EMRELIS and for 1 month after the last dose.
8.3 Females and Males of Reproductive Potential
EMRELIS can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1 ) ] .
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating EMRELIS treatment.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with EMRELIS and for 2 months after the last dose.
Males
Based on genotoxicity findings, advise male patients with female partners of reproductive potential to use effective contraception during treatment with EMRELIS and for 4 months after the last dose.
Infertility
Females
Based on findings in animal studies with MMAE-containing antibody-drug conjugates (ADCs), EMRELIS may impair female fertility. The effect on fertility is reversible [see Nonclinical Toxicology (13.1 )] .
Males
Based on findings from animal studies, EMRELIS may impair male fertility. The reversibility of this effect is unknown [see Nonclinical Toxicology (13.1 )] .
8.4 Pediatric Use
Safety and effectiveness of EMRELIS have not been established in pediatric patients.
8.5 Geriatric Use
Of the 168 patients with previously treated EGFR wild-type non-squamous NSCLC with c-Met protein overexpression treated with EMRELIS in LUMINOSITY, 50% were ≥65 years of age and 12% were ≥75 years of age. No overall differences in safety or effectiveness were observed between older and younger patients.
8.6 Hepatic Impairment
Avoid use of EMRELIS in patients with moderate or severe hepatic impairment (total bilirubin >1.5 x ULN and any AST).
Patients with moderate or severe hepatic impairment are likely to have increased exposure to MMAE, which may increase the risk of adverse reactions. EMRELIS has not been studied in patients with moderate or severe hepatic impairment.
No dosage adjustment is recommended for patients with mild hepatic impairment (total bilirubin ≤ULN and AST >ULN or total bilirubin >ULN and ≤1.5 x ULN and any AST) [see Clinical Pharmacology (12.3 ) ] .
4 CONTRAINDICATIONS
None.
5 WARNINGS AND PRECAUTIONS
- Peripheral Neuropathy : Monitor patients for new or worsening peripheral neuropathy. Withhold, reduce the dose, or permanently discontinue EMRELIS based on the severity. (5.1 )
- I nterstitial Lung Disease (ILD)/ Pneumonitis : Severe, life-threatening or fatal ILD/pneumonitis may occur. Withhold or permanently discontinue EMRELIS based on the severity. (5.2 )
- Ocular Surface Disorders : Monitor patients for signs or symptoms of ocular surface disorders, including vision changes. Withhold or permanently discontinue EMRELIS based on the severity. (5.3 )
- Infusion - Related Reactions (IRR) : Monitor patients for IRR. Withhold, reduce the rate of infusion, or permanently discontinue EMRELIS based on the severity. For patients who experience IRR, administer premedications prior to subsequent infusions. (5.4 )
- Embryo-Fetal Toxicity : Can cause fetal harm. Advise patients about the potential risk to a fetus and to use effective contraception. (5.5 )
5.1 Peripheral Neuropathy
EMRELIS can cause peripheral neuropathy, including peripheral sensory neuropathy and peripheral motor neuropathy.
In the safety population [see Adverse Reactions (6.1 )], peripheral neuropathy occurred in 51% of patients treated with EMRELIS, including Grade 3 in 11%. These adverse reactions included peripheral sensory neuropathy in 45% of patients and peripheral motor neuropathy in 9%. The median time to onset of peripheral neuropathy was 105 days (range: 1 to 472 days). Peripheral neuropathy led to permanent discontinuation of EMRELIS in 13% of patients. The median time to onset of peripheral neuropathy leading to treatment discontinuation was 249 days (range: 57 to 519 days). Of the 7 patients with motor neuropathy ongoing as of their last dose of EMRELIS, 6 had persistent Grade 1 or 2 symptoms 30 days after their last dose.
Monitor patients for signs and symptoms of new or worsening peripheral neuropathy such as hypoesthesia, hyperesthesia, paresthesia, a burning sensation, neuropathic pain, or muscle weakness. Withhold, reduce the dose or permanently discontinue EMRELIS based on severity [see Dosage and Administration (2.3 )].
5.2 Interstitial Lung Disease /Pneumonitis
EMRELIS can cause severe, life-threatening, or fatal interstitial lung disease (ILD)/pneumonitis.
In the safety population [see Adverse Reactions (6.1 )], ILD/pneumonitis occurred in 10% of patients treated with EMRELIS, including Grade 3 in 3% and Grade 4 in 0.6%. There were 3 fatal cases of ILD/pneumonitis in patients who received EMRELIS. The median time to onset of ILD/pneumonitis was 48 days (range: 23 to 85 days). ILD/pneumonitis led to permanent discontinuation of EMRELIS in 7% of patients. The median time to onset of ILD/pneumonitis leading to treatment discontinuation was 46 days (range: 23 to 85 days).
Advise patients to immediately report cough, dyspnea, fever, and/or any new or worsening respiratory symptoms. Monitor patients for signs and symptoms of ILD/pneumonitis. Withhold or permanently discontinue EMRELIS based on severity [see Dosage and Administration (2.3 )].
5.3 Ocular Surface Disorders
EMRELIS can cause ocular surface disorders including blurred vision, visual impairment, keratitis, and dry eye.
In the safety population [see Adverse Reactions (6.1 )], ocular surface disorders occurred in 25% of patients treated with EMRELIS. The most common ocular surface disorders were blurred vision (15%), keratitis (11%), and dry eye (5%). Grade 3 ocular surface disorders occurred in 1.2% of patients [blurred vision (1.2%), and keratitis (0.6%)]. The median time to onset of ocular surface disorders was 47 days (range: 1 to 319 days).
Monitor patients for ocular surface disorders during treatment with EMRELIS. Withhold EMRELIS and refer patients to an eye care professional for an ophthalmic examination and treatment for patients who develop Grade ≥2 ocular toxicity. Withhold or permanently discontinue EMRELIS based on severity [see Dosage and Administration (2.3 )].
5.4 Infusion - Related Reactions
EMRELIS can cause infusion-related reactions (IRR); signs and symptoms of IRR include dyspnea, flushing, chills, nausea, chest discomfort, and hypotension. The median time to onset of IRR was 28 days (range: 1 to 43 days).
In the safety population, [see Adverse Reactions (6.1 )] , IRR occurred in 3% of patients treated with EMRELIS including Grade 3 in 1.2% and Grade 4 in 0.6%. IRR led to permanent discontinuation of EMRELIS in 0.6% of patients.
Monitor patients for signs and symptoms of infusion reactions during EMRELIS infusion. Withhold, reduce the rate of infusion, or permanently discontinue EMRELIS based on severity [see Dosage and Administration (2.3 )]. For patients who experience IRR, administer premedications prior to subsequent infusions.
5.000000000000000e+00 5 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, EMRELIS can cause fetal harm when administered to a pregnant woman. The small molecule component of EMRELIS, MMAE, administered to rats caused adverse developmental outcomes, including embryo-fetal mortality and structural abnormalities, at exposures similar to those occurring clinically at the recommended dose.
Advise patients of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with EMRELIS and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with EMRELIS and for 4 months after the last dose [see Use in Specific Populations (8.1 , 8.3 ) and Clinical Pharmacology (12.1 )].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
LUMINOSITY
The safety population described in WARNINGS AND PRECAUTIONS and below reflects exposure to EMRELIS in 168 patients with locally advanced or metastatic EGFR wild-type non-squamous NSCLC with c-Met protein overexpression who received EMRELIS as a single agent administered at 1.9 mg/kg intravenously every 2 weeks in the LUMINOSITY study [see Clinical Studies (14 )]. Among patients who received EMRELIS, 42% were exposed for 6 months or longer and 11% were exposed for greater than one year.
The median age of patients who received EMRELIS was 64.5 years (range: 33 to 83 years); 70% were male; 65% were White; 1.8% were Black or African American, 33% were Asian; and 0.6% were of Hispanic or Latino ethnicity.
Serious adverse reactions occurred in 35% of patients. Serious adverse reactions occurring in ≥2% of patients included ILD/pneumonitis (5%), pneumonia (5%), peripheral neuropathy (3.6%), and pleural effusion (2.4%). Fatal adverse reactions occurred in 5% of patients who received EMRELIS, including ILD/pneumonitis (1.8%), pneumonia (1.2%), sudden death (1.2%), noninfectious endocarditis (0.6%) and myocardial infarction (0.6%).
Permanent discontinuations of EMRELIS due to adverse reactions occurred in 30% of patients. Adverse reactions which resulted in permanent discontinuation of EMRELIS in ≥2% included peripheral neuropathy and ILD/pneumonitis.
Dosage interruptions due to adverse reactions occurred in 44% of patients. Adverse reactions which required dosage interruption in ≥2% of patients included peripheral neuropathy, fatigue, pneumonia, increased ALT, blurred vision, COVID-19, ILD/pneumonitis, and keratitis.
Dose reductions due to adverse reactions occurred in 28% of patients. Adverse reactions which required dose reductions in ≥2% of patients included peripheral neuropathy, fatigue, and keratitis.
The most common adverse reactions (≥20%) were peripheral neuropathy, fatigue, decreased appetite, and peripheral edema.
The most common Grade 3 or 4 laboratory abnormalities (≥2%) were decreased lymphocytes, increased glucose, increased ALT, increased gamma glutamyl transferase, decreased phosphorus, decreased sodium, decreased hemoglobin and decreased calcium.
Table 5 summarizes the adverse reactions in LUMINOSITY.
| Adverse Reaction | EMRELIS (N=168) | |
| All Grades 1 % | Grade 3 or 4 1 % | |
| Nervous system disorders | ||
| Peripheral neuropathy 2 | 51 | 11 |
| General disorders and administration site conditions | ||
| Fatigue 2 | 29 | 3.6 |
| Peripheral edema 2 | 22 | 1.8 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 22 | 0.6 |
| Gastrointestinal disorders | ||
| Nausea | 15 | 0 |
| Constipation | 14 | 0.6 |
| Vomiting | 10 | 0.6 |
| Eye disorders | ||
| Blurred vision 3 | 15 | 1.2 |
| Keratitis 4 | 11 | 0.6 |
| Infections and infestations | ||
| Pneumonia 2 | 13 | 6 |
| Respiratory, thoracic and mediastinal disorders | ||
| ILD/pneumonitis 2 | 10 | 3.6 |
| 1 Events were graded using NCI CTCAE version 4.03. 2 Grouped term. 3 Includes vision blurred, visual acuity reduced, visual impairment. 4 Includes corneal cyst, corneal disorder, corneal erosion, corneal edema, corneal opacity, keratitis, keratitis interstitial, punctate keratitis. | ||
Other clinically relevant adverse reactions in <10% of patients who received EMRELIS included arthralgia, dizziness, dry eye, infusion-related reaction and photophobia.
Table 6 presents laboratory abnormalities in LUMINOSITY.
| Laboratory Abnormality | EMRELIS (N=168) | |
| All Grades % | Grade 3 or 4 % | |
| Chemistry | ||
| Albumin decreased | 61 | 0.6 |
| Glucose increased | 58 | 4.8 |
| Calcium decreased | 47 | 2.4 |
| Alanine transaminase increased | 41 | 4.8 |
| Gamma glutamyl transferase increased | 36 | 4.3 |
| Aspartate aminotransferase increased | 34 | 0.6 |
| Phosphorus decreased | 33 | 4.2 |
| Sodium decreased | 30 | 3.6 |
| Alkaline phosphatase increased | 30 | 0.6 |
| Creatinine increased | 16 | 1.2 |
| Potassium decreased | 14 | 1.2 |
| Magnesium decreased | 14 | 0.6 |
| Glucose decreased | 11 | 0 |
| Magnesium increased | 10 | 0 |
| Hematology | ||
| Lymphocytes decreased | 37 | 10 |
| Hemoglobin decreased | 35 | 3.6 |
| White blood cells decreased | 16 | 1.2 |
| Platelets decreased | 14 | 0.6 |
| Neutrophils decreased | 10 | 1.2 |
Percentages were calculated using patients with worsening laboratory values from baseline and the number of patients with both baseline and post-treatment measurements as the denominator.
7 DRUG INTERACTIONS
S trong CYP3A I nhibitors : concomitant use with EMRELIS may increase the AUC of MMAE. Monitor for increased risk of adverse reactions to EMRELIS. (7.1 )
7.1 Effect of Other Drugs on EMRELIS
Strong CYP3A Inhibitors
Concomitant use with strong CYP3A inhibitors may increase unconjugated MMAE AUC [see Clinical Pharmacology (12.3 ) ] , which may increase the risk of EMRELIS adverse reactions. Monitor patients for adverse reactions when EMRELIS is given concomitantly with strong CYP3A inhibitors.
11 DESCRIPTION
Telisotuzumab vedotin-tllv is a c-Met directed antibody-drug conjugate (ADC) comprised of a humanized immunoglobulin G1 kappa (IgG1κ) monoclonal antibody conjugated to the small molecule microtubule-disrupting agent, monomethyl auristatin E (MMAE), via a protease-cleavable valine-citrulline (vc) linker. The antibody is produced in a mammalian cell line (Chinese hamster ovary) and the drug-linker is produced by chemical synthesis. Each monoclonal antibody molecule carries an average of 3 MMAE molecules. Telisotuzumab vedotin-tllv has an approximate molecular weight of 152 kDa.
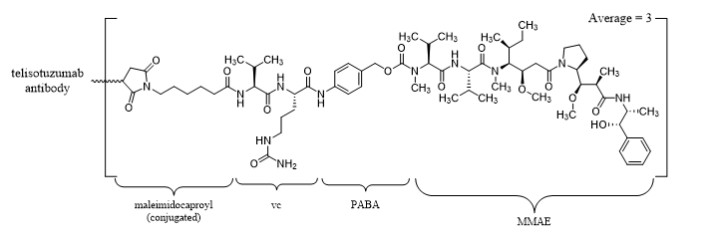
EMRELIS (telisotuzumab vedotin-tllv) for injection is a sterile, white to off-white, preservative-free, lyophilized powder in a single-dose vial for reconstitution and dilution prior to intravenous infusion. EMRELIS is supplied as 20 mg per vial or 100 mg per vial and requires reconstitution with Sterile Water for Injection, USP (1.1 mL and 5.2 mL, respectively) to obtain a concentration of 20 mg/mL [see Dosage and Administration (2.4 )] . Following reconstitution, each mL delivers 20 mg of telisotuzumab vedotin-tllv, and histidine (2.33 mg), polysorbate 80 (0.10 mg), sucrose (70.0 mg), and Water for Injection. Hydrochloric acid was added to adjust the pH to 6.0.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Telisotuzumab vedotin-tllv is a c-Met-directed antibody drug conjugate (ADC). The antibody is a humanized IgG1κ directed against c-Met, the cell surface receptor for hepatocyte growth factor. The small molecule, MMAE, is a microtubule-disrupting agent, attached to the antibody via a protease cleavable linker. Following binding to c-Met-expressing cells, telisotuzumab vedotin-tllv undergoes internalization and intracellular cleavage of MMAE. MMAE disrupts the microtubule network of actively dividing cells, subsequently inducing cell cycle arrest and apoptotic cell death. Telisotuzumab vedotin-tllv exhibited antitumor activity in xenograft models of NSCLC.
12.2 Pharmacodynamics
Exposure Response Relationships
Exposure-response relationships for efficacy and time course of pharmacodynamic response have not been fully characterized.
Higher telisotuzumab vedotin-tllv exposure was associated with increased rates of Grade 3 peripheral neuropathy, and Grades 2 and 3 ocular surface disorders. Higher unconjugated MMAE exposure was associated with increased rates of Grade ≥3 adverse reactions and fatal adverse reactions.
Cardiac Electrophysiology
There is insufficient information to characterize the effect of telisotuzumab vedotin-tllv on the QT interval.
12.3 Pharmacokinetics
The exposure parameters of telisotuzumab vedotin-tllv (ADC) and unconjugated MMAE (cytotoxic component of EMRELIS) are summarized in Table 7. The plasma exposure of the ADC and unconjugated MMAE increased proportionally over a dose range of 1.2 to 3.3 mg/kg (0.64 to 1.7 times the approved recommended dose). ADC time to maximum plasma concentrations (Tmax) occurs at the end of intravenous infusion while MMAE Tmax occurs approximately 5 days after EMRELIS administration. Minimal accumulation of the ADC or MMAE occurs.
| ADC | Unconjugated MMAE | |
| C max | 29 (43) µg/mL | 2.2 (53) ng/mL |
| AUC 0- tau | 2,130 (55) µg∙hr/mL | 405 (64) ng∙hr/mL |
C max = maximum concentration
AUC 0- tau = area under the concentration-time curve from time zero to 14 days (2 weeks)
Distribution
The estimated volume of distribution is 3.4 L (16.5%CV) for telisotuzumab vedotin-tllv.
MMAE plasma protein binding ranges from 68% to 82% in vitro .
Elimination
The elimination half-life is approximately 3 days for the ADC and approximately 4 days for MMAE. The estimated clearance (CL) is 1.3 L/day (31.5%CV) for the ADC and 76 L/day (52.1%CV) for MMAE.
Metabolism
Telisotuzumab vedotin-tllv catabolism has not been studied in humans; however, it is expected to undergo catabolism to small peptides, amino acids, unconjugated MMAE, and unconjugated MMAE-related catabolites. MMAE is metabolized primarily by CYP3A4.
Specific Populations
No clinically significant differences in the pharmacokinetics of the ADC or unconjugated MMAE were observed based on age (30 to 87 years), sex, race (65% White, 32% Asian, and 3% Black), body weight (34 to 144 kg), mild or moderate renal impairment (CLcr 30 to 89 mL/min, estimated by Cockcroft-Gault), or mild hepatic impairment (total bilirubin ≤ULN and AST >ULN or total bilirubin >ULN and ≤1.5 x ULN and any AST).
The effect of severe renal impairment (CLcr <30 mL/min), end-stage renal disease with or without dialysis, or moderate to severe hepatic impairment (total bilirubin >1.5 × ULN and any AST) on the pharmacokinetics of telisotuzumab vedotin-tllv or unconjugated MMAE is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches:
No clinical studies have evaluated the potential for drug-drug interactions.
Strong CYP3A4 Inhibitor:
MMAE AUC is predicted to increase by 1.4-fold following concomitant administration of EMRELIS with ketoconazole (a strong CYP3A inhibitor).
Strong CYP3A4 Inducer:
MMAE AUC is predicted to decrease by 70% following concomitant administration of EMRELIS with rifampicin (a strong CYP3A inducer).
Sensitive CYP3A Substrate:
No clinically significant difference in the pharmacokinetics of midazolam (a sensitive CYP3A substrate) is predicted when used concomitantly with EMRELIS.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes:
MMAE is a substrate and an inhibitor of CYP3A4/5.
Transporter Systems:
MMAE is a substrate of P-glycoprotein (P-gp).
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the study described below with the incidence of anti-drug antibodies in other studies, including those of EMRELIS or of other telisotuzumab products.
Following administration of EMRELIS in LUMINOSITY, 22% (60/269) of patients tested positive for treatment-emergent antibodies against telisotuzumab vedotin-tllv at one or more post-baseline time points. Of those who tested positive for anti-drug antibody, neutralizing antibodies were detected in 38% (23/60) of patients. Development of anti-drug antibodies and neutralizing antibodies resulted in a 17% decrease in clearance of telisotuzumab vedotin-tllv. There are insufficient data to assess whether the observed anti-telisotuzumab vedotin-tllv antibody-associated pharmacokinetic changes affect the safety and effectiveness of EMRELIS.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies in animals have not been performed with telisotuzumab vedotin-tllv or the small molecule MMAE.
Mutagenesis
MMAE was positive for genotoxicity in the in vivo rat bone marrow micronucleus study through an aneugenic mechanism. MMAE was not mutagenic in the bacterial reverse mutation (Ames) assay or the L5178Y TK +/- mouse lymphoma forward mutation assay.
Impairment of Fertility
Fertility studies with telisotuzumab vedotin-tllv or MMAE have not been conducted. However, results of repeat-dose toxicity studies in rats and monkeys indicate the potential for telisotuzumab vedotin-tllv to impair male and female reproductive function and fertility.
In a 2-week repeat-dose toxicology study in rats, telisotuzumab vedotin-tllv administered at doses ≥6 mg/kg (≥12 times the human exposure [AUC] at the recommended dose) resulted in decreased number/degeneration of germ cells in the testes largely due to loss of spermatogonia. The reversibility of these findings was not assessed.
In a 4-week repeat-dose toxicology study in sexually immature monkeys, telisotuzumab vedotin-tllv administered at doses ≥3 mg/kg (≥4 times the human exposure [AUC] at the recommended dose) resulted in degeneration of granulosa cells and decreased number of tertiary follicles in the ovaries and degeneration/necrosis of endometrial glands in the uterus. There was evidence of reversibility after an 8-week recovery period.
14 CLINICAL STUDIES
Previously Treated EGFR Wild-Type N on-squamous NSCLC with High c-Met Protein Overexpression
LUMINOSITY
The efficacy of EMRELIS was evaluated in the LUMINOSITY study (NCT03539536), a multicenter, open-label, single-arm, multi-cohort clinical trial. Eligible patients were required to have locally advanced or metastatic NSCLC with c-Met protein overexpression and treatment with prior systemic therapy (including no more than one line of prior chemotherapy) in the locally advanced or metastatic setting. The study excluded patients who had received radiation therapy to the lungs <6 months prior to enrollment and patients who had a history of ILD/pneumonitis requiring treatment with steroids or ILD/pneumonitis within 3 months of the first dose.
Patients received EMRELIS at 1.9 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity. The major efficacy outcome measure was overall response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 as assessed by a blinded independent central review (BICR). An additional efficacy outcome measure was duration of response (DOR) by BICR.
The efficacy population included 84 patients with non-squamous, EGFR wild-type NSCLC with high c-Met protein overexpression who had received prior systemic therapy. High c-Met protein overexpression was defined as ≥50% of tumor cells with strong (3+) membrane staining on archival or recent tissue samples by immunohistochemistry (IHC) and was determined by prospective testing at a central laboratory prior to enrollment using the MET (SP44) clinical trial assay (CTA). Of the 84 patients with high c-Met protein overexpression identified by central testing using the CTA, tissue samples from 38/84 (45%) patients were tested retrospectively using the VENTANA MET (SP44) RxDx assay. One sample was unevaluable. Of the 37 samples retested and evaluable, 32 (87%) samples were confirmed to have high c-Met protein overexpression, defined as ≥50% of tumor cells with strong (3+) membrane and/or cytoplasmic staining.
The median age was 64 years (range: 38 to 83 years); 75% were male; 61% were White, 1.2% were Black or African American, 38% were Asian; none were of Hispanic or Latino ethnicity. Twenty-five percent had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 and 74% had ECOG PS of 1; 19% were never smokers, 68% were former smokers, and 13% were current smokers; 99% had Stage IV disease; and 19% of patients had previously treated brain metastases. The median number of lines of prior therapies was 1 (range 1 - 3); 73% of patients had one line, 24% had two lines, and 3.6% had three lines of prior systemic therapy; 96% of patients had prior platinum therapy, 82% had prior immunotherapy (anti-PD-1/PD-L1), 6% had prior targeted therapy, and 3.6% had prior MET tyrosine kinase inhibitor therapy.
Efficacy results are summarized in Table 8.
| Efficacy Parameter | EMRELIS N = 84 |
| Confirmed Overall Response Rate (ORR) , % (95% CI) | 35 (24, 46) |
| Complete Response, % | 0 |
| Partial Response, % | 35 |
| Duration of Response | N = 29 |
| Median, months (95% CI) | 7.2 (4.2, 12) |
| DOR ≥6 months, a % | 59 |
| DOR ≥12 months, a % | 21 |
| CI=confidence interval; DOR=duration of response a Based on observed duration of response in 29 responders. | |
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
EMRELIS (telisotuzumab vedotin-tllv) for injection is a sterile, preservative-free, white to off-white lyophilized powder, supplied in a glass single-dose vial.
- Carton of one 20 mg/vial (NDC 0074-1044-01)
- Carton of one 100 mg/vial (NDC 0074-1055-01)
Storage and Handling
Store refrigerated at 2 o C to 8 o C (36 o F to 46 o F) in original carton to protect from light. Do not freeze. Do not shake.
Special Handling
EMRELIS is a hazardous product. Follow special handling and disposal procedures. 1
12.1 Mechanism of Action
Telisotuzumab vedotin-tllv is a c-Met-directed antibody drug conjugate (ADC). The antibody is a humanized IgG1κ directed against c-Met, the cell surface receptor for hepatocyte growth factor. The small molecule, MMAE, is a microtubule-disrupting agent, attached to the antibody via a protease cleavable linker. Following binding to c-Met-expressing cells, telisotuzumab vedotin-tllv undergoes internalization and intracellular cleavage of MMAE. MMAE disrupts the microtubule network of actively dividing cells, subsequently inducing cell cycle arrest and apoptotic cell death. Telisotuzumab vedotin-tllv exhibited antitumor activity in xenograft models of NSCLC.