Get your patient on Fetroja (Cefiderocol Sulfate Tosylate)
Dosage & administration

Fetroja prescribing information
INDICATIONS AND USAGE
FETROJA is a cephalosporin antibacterial indicated in patients 18 years of age or older for the treatment of the following infections caused by susceptible Gram-negative microorganisms:
- Complicated Urinary Tract Infections (cUTI), including Pyelonephritis (1.1 )
- Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP) (1.2 )
To reduce the development of drug-resistant bacteria and maintain the effectiveness of FETROJA and other antibacterial drugs, FETROJA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1.3 )
Complicated Urinary Tract Infections (cUTIs), Including Pyelonephritis
FETROJA ® is indicated in patients 18 years of age or older for the treatment of complicated urinary tract infections (cUTIs), including pyelonephritis caused by the following susceptible Gram-negative microorganisms: Escherichia coli , Klebsiella pneumoniae , Proteus mirabilis , Pseudomonas aeruginosa , and Enterobacter cloacae complex [see Clinical Studies (14.1) ] .
Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
FETROJA is indicated in patients 18 years of age or older for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia, caused by the following susceptible Gram-negative microorganisms: Acinetobacter baumannii complex, Escherichia coli , Enterobacter cloacae complex, Klebsiella pneumoniae , Pseudomonas aeruginosa , and Serratia marcescens [see Clinical Studies (14.2) ].
Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of FETROJA and other antibacterial drugs, FETROJA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
DOSAGE AND ADMINISTRATION
- Administer 2 grams of FETROJA for injection every 8 hours by intravenous (IV) infusion over 3 hours in patients with creatinine clearance (CLcr) 60 to 119 mL/min. (2.1 )
- Dose adjustments are required for patients with CLcr less than 60 mL/min, (including patients receiving intermittent hemodialysis (HD) or continuous renal replacement therapy (CRRT)), and for patients with CLcr 120 mL/min or greater. (2.2 )
- See full prescribing information for instructions on preparation of FETROJA doses. (2.3 )
- See full prescribing information for drug compatibilities. (2.4 )
Recommended Dosage
The recommended dosage of FETROJA is 2 grams administered every 8 hours by intravenous (IV) infusion over 3 hours in adults with a creatinine clearance (CLcr) of 60 to 119 mL/min.
Dosage adjustment of FETROJA is recommended for patients with CLcr less than 60 mL/min, including patients receiving intermittent hemodialysis (HD) or continuous renal replacement therapy (CRRT), and for patients with CLcr 120 mL/min or greater [see Dosage and Administration (2.2) ]. The recommended duration of treatment with FETROJA is 7 to 14 days. The duration of therapy should be guided by the patient's clinical status.
Dosage Adjustments in Patients with CLcr Less Than 60 mL/min (Including Patients Undergoing Intermittent HD or CRRT), and CLcr 120 mL/min or Greater
Dosage Adjustments in Patients with CLcr Less Than 60 mL/min Including Patients Receiving Intermittent HD
Dosage adjustment of FETROJA is recommended in patients with CLcr less than 60 mL/min (Table 1). For patients undergoing intermittent HD, start the dosing of FETROJA immediately after the completion of HD. For patients with fluctuating renal function, monitor CLcr and adjust dosage accordingly.
| Estimated Creatinine Clearance (CLcr) CLcr = creatinine clearance estimated by Cockcroft-Gault equation. | Dose | Frequency | Infusion Time |
|---|---|---|---|
| HD = hemodialysis. | |||
| CLcr 30 to 59 mL/min | 1.5 grams | Every 8 hours | 3 hours |
| CLcr 15 to 29 mL/min | 1 gram | Every 8 hours | 3 hours |
| CLcr less than 15 mL/min, with or without intermittent HD Cefiderocol is removed by HD; administer FETROJA immediately after HD for patients receiving intermittent HD. | 0.75 grams | Every 12 hours | 3 hours |
Dosage Adjustments in Patients Receiving CRRT
For patients receiving CRRT, including continuous venovenous hemofiltration (CVVH), continuous venovenous hemodialysis (CVVHD), and continuous venovenous hemodiafiltration (CVVHDF), the dosage of FETROJA should be based on the effluent flow rate in CRRT (see Table 2 ). These recommendations are intended to provide initial dosing in patients receiving CRRT. Dosing regimens may need to be tailored based on residual renal function and patient's clinical status [see Use in Specific Populations (8.6) ] .
| Effluent Flow Rate Ultrafiltrate flow rate for CVVH, dialysis flow rate for CVVHD, ultrafiltrate flow rate plus dialysis flow rate for CVVHDF. | Recommended Dosage of FETROJA |
|---|---|
| CRRT = continuous renal replacement therapy. | |
| 2 L/hr or less | 1.5 grams every 12 hours |
| 2.1 to 3 L/hr | 2 grams every 12 hours |
| 3.1 to 4 L/hr | 1.5 grams every 8 hours |
| 4.1 L/hr or greater | 2 grams every 8 hours |
Dosage Adjustments in Patients with CLcr 120 mL/min or Greater
For patients with CLcr greater than or equal to 120 mL/min, FETROJA 2 grams administered every 6 hours by IV infusion over 3 hours is recommended [see Use in Specific Populations (8.6) ] .
Preparation of FETROJA Solution for Administration
FETROJA is supplied as a sterile, lyophilized powder that must be reconstituted and subsequently diluted using aseptic technique prior to intravenous infusion.
Preparation of Doses
Reconstitute the powder for injection in the FETROJA vial with 10 mL of either 0.9% sodium chloride injection, USP or 5% dextrose injection, USP and gently shake to dissolve. Allow the vial(s) to stand until the foaming generated on the surface has disappeared (typically within 2 minutes). The reconstituted solution will have a final volume of approximately 11.2 mL and concentration of 0.089 gram/mL. The reconstituted solution is for intravenous infusion only after dilution in an appropriate infusion solution.
To prepare the required doses, withdraw the appropriate volume of reconstituted solution from the vial according to Table 3 below. Add the withdrawn volume to a 100 mL infusion bag containing 0.9% sodium chloride injection, USP or 5% dextrose injection, USP [see Dosage and Administration (2.4) ] .
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. FETROJA infusions are clear, colorless solutions. Discard any unused FETROJA solution in the vial (see Table 3 ).
| FETROJA Dose | Number of 1-gram FETROJA Vials to be Reconstituted | Volume to Withdraw from Reconstituted Vial(s) | Total Volume of FETROJA Reconstituted Solution for Further Dilution into a 100 mL Infusion Bag |
|---|---|---|---|
| 2 grams | 2 vials | 11.2 mL (entire contents) of each vial | 22.4 mL |
| 1.5 grams | 2 vials | 11.2 mL (entire contents) of first vial AND 5.6 mL from second vial | 16.8 mL |
| 1 gram | 1 vial | 11.2 mL (entire contents) | 11.2 mL |
| 0.75 gram | 1 vial | 8.4 mL | 8.4 mL |
Drug Compatibility
FETROJA solution for administration is compatible with:
- 0.9% sodium chloride injection, USP
- 5% dextrose injection, USP
The compatibility of FETROJA solution for administration with solutions containing other drugs or other diluents has not been established.
Storage of Reconstituted Solutions
Reconstituted FETROJA
Upon reconstitution with the appropriate diluent, the reconstituted FETROJA solution in the vial should be immediately transferred and diluted into the infusion bag. Reconstituted FETROJA can be stored for up to 1 hour at room temperature in the vial. Discard any unused reconstituted solution.
Diluted FETROJA Infusion Solution
The diluted FETROJA infusion solution in the infusion bag is stable for up to 6 hours at room temperature.
The diluted FETROJA infusion solution in the infusion bag may also be refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours, protected from light; and then the infusion should be completed within 6 hours at room temperature.
DOSAGE FORMS AND STRENGTHS
FETROJA 1 gram for injection is supplied as a white to off-white, sterile, lyophilized powder for reconstitution in single-dose, clear glass vials; each vial contains 1 gram of cefiderocol.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
There are no available data on FETROJA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
Available data from published prospective cohort studies, case series, and case reports over several decades with cephalosporin use in pregnant women have not established drug-associated risks of major birth defects, miscarriage, or adverse maternal or fetal outcomes (see Data ) .
Developmental toxicity studies with cefiderocol administered during organogenesis to rats and mice showed no evidence of embryo-fetal toxicity, including drug-induced fetal malformations, at doses providing exposure levels 0.9 times (rats) or 1.3 times (mice) higher than the average observed in patients receiving the maximum recommended daily dose.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
While available studies cannot definitively establish the absence of risk, published data from prospective cohort studies, case series, and case reports over several decades have not identified an association with cephalosporin use during pregnancy and major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Available studies have methodologic limitations, including small sample size, retrospective data collection, and inconsistent comparator groups.
Animal Data
Developmental toxicity was not observed in rats at intravenous doses of up to 1000 mg/kg/day or mice at subcutaneous doses of up to 2000 mg/kg/day given during the period of organogenesis (gestation days 6-17 in rats and 6-15 in mice). No treatment-related malformations or reductions in fetal viability were observed. Mean plasma exposure (AUC) at these doses was approximately 0.9 times (rats) and 1.3 times (mice) the daily mean plasma exposure in patients that received 2 grams of cefiderocol infused intravenously every 8 hours.
In a pre- and postnatal development study, cefiderocol was administered intravenously at doses up to 1000 mg/kg/day to rats from Day 6 of pregnancy until weaning. No adverse effects on parturition, maternal function, or pre- and postnatal development and viability of the pups were observed.
In pregnant rats, cefiderocol-derived radioactivity was shown to cross the placenta, but the amount detected in fetuses was a small percentage (< 0.5%) of the dose.
Lactation
Risk Summary
It is not known whether cefiderocol is excreted into human milk; however, cefiderocol-derived radioactivity was detected in the milk of lactating rats that received the drug intravenously. When a drug is present in animal milk, it is likely that the drug will be present in human milk. No information is available on the effects of FETROJA on the breastfed infant or on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for FETROJA and any potential adverse effects on the breastfed child from FETROJA or from the underlying maternal condition.
Data
Cefiderocol-derived radioactivity was detected in milk following intravenous administration to lactating rats. The peak level in rat milk was approximately 6% of the peak plasma level.
Pediatric Use
Safety and effectiveness of FETROJA in pediatric patients younger than 18 years of age have not been established.
Geriatric Use
cUTI
Of the 300 patients treated with FETROJA in the cUTI trial, 158 (52.7%) were 65 years of age and older, and 67 (22.3%) were 75 years of age and older. No overall differences in safety or efficacy were observed between these patients and younger patients.
HABP/VABP
Of the 148 patients treated with FETROJA in the HABP/VABP trial, 83 (56.1%) were 65 years of age and older, and 40 (27%) were 75 years of age and older.
The incidence of adverse reactions in patients treated with FETROJA was similar in patients under 65 years of age as compared to older patients (65 years of age and older and 75 years of age and older). The incidence of adverse reactions in older patients (65 years of age and older and 75 years of age and older) was also similar between treatment groups.
Clinical cure rates at the Test-of-Cure visit (TOC) in FETROJA-treated adult patients younger than 65 years of age, 65 years of age to younger than 75 years of age, and 75 years of age and older were 60%, 77.5%, and 60%, respectively. In comparison, the clinical cure rates at the TOC visit in the meropenem-treated patients for each of these subgroups were 65.5%, 64.4%, and 70.5%, respectively. The observed all-cause mortality rates at Day 14 in the FETROJA-treated patients for each of these subgroups were 12.3%, 7.5%, and 17.5%, respectively. In comparison, in the meropenem-treated patients for each of these subgroups, they were 10.3%, 17.8%, and 9.1%, respectively.
cUTI and HABP/VABP
FETROJA is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. No dosage adjustment is required based on age. Dosage adjustment for elderly patients should be based on renal function [see Dosage and Administration (2.2) , Use in Specific Populations (8.6) , and Clinical Pharmacology (12.3) ] .
Renal Impairment
Patients with CLcr 60 to 89 mL/min
No dosage adjustment of FETROJA is recommended in patients with CLcr 60 to 89 mL/min.
Patients with CLcr Less Than 60 mL/min Including Patients Receiving Intermittent HD
Dose adjustment is required in patients with CLcr less than 60 mL/min, and in patients who are receiving HD. In patients requiring HD, complete HD at the latest possible time before the start of cefiderocol dosing [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3) ] . Monitor renal function regularly and adjust the dosage of FETROJA accordingly as renal function may change during the course of therapy.
Patients Receiving CRRT
A total of 16 patients treated with FETROJA received CRRT in clinical trials. Dosage adjustment of FETROJA is required in patients receiving CRRT including CVVH, CVVHD, and CVVHDF. Dosage of FETROJA should be based on the effluent flow rate in patients receiving CRRT [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3) ]. While on CRRT, a patient's residual renal function may change. Improvements or reductions in residual renal function may warrant a change in FETROJA dosage.
Patients with CLcr 120 mL/min or Greater
CLcr 120 mL/min or greater may be seen in seriously ill patients, who are receiving intravenous fluid resuscitation. Dosage adjustment of FETROJA is required in patients with CLcr 120 mL/min or greater [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3) ]. Monitor renal function regularly and adjust the dosage of FETROJA accordingly as renal function may change during the course of therapy.
Hepatic Impairment
The effects of hepatic impairment on the pharmacokinetics of cefiderocol have not been evaluated. Hepatic impairment is not expected to alter the elimination of cefiderocol as hepatic metabolism/excretion represents a minor pathway of elimination for cefiderocol. Dosage adjustments are not necessary in patients with impaired hepatic function.
CONTRAINDICATIONS
FETROJA is contraindicated in patients with a known history of severe hypersensitivity to cefiderocol or other beta-lactam antibacterial drugs, or any other component of FETROJA [see Warnings and Precautions (5.2) and Adverse Reactions (6.1) ] .
WARNINGS AND PRECAUTIONS
- Increase in All-Cause Mortality in Patients with Carbapenem-Resistant Gram-Negative Bacterial Infections: An increase in all-cause mortality was observed in FETROJA-treated patients compared to those treated with best available therapy (BAT). Closely monitor the clinical response to therapy in patients with cUTI and HABP/VABP. (5.1 )
- Hypersensitivity Reactions: Serious and occasionally fatal hypersensitivity (anaphylactic) reactions have been reported in patients receiving beta-lactam antibacterial drugs. Hypersensitivity was observed with FETROJA. Cross-hypersensitivity may occur in patients with a history of penicillin allergy. If an allergic reaction occurs, discontinue FETROJA. (5.2 )
- Clostridioides difficile -associated Diarrhea (CDAD): CDAD has been reported with nearly all systemic antibacterial agents, including FETROJA. Evaluate if diarrhea occurs. (5.3 )
- Seizures and Other Central Nervous System (CNS) Adverse Reactions: CNS adverse reactions such as seizures have been reported with FETROJA. If focal tremors, myoclonus, or seizures occur, evaluate patients to determine whether FETROJA should be discontinued. (5.4 )
Increase in All-Cause Mortality in Patients with Carbapenem-Resistant Gram-Negative Bacterial Infections
An increase in all-cause mortality was observed in patients treated with FETROJA as compared to best available therapy (BAT) in a multinational, randomized, open-label trial in critically ill patients with carbapenem-resistant Gram-negative bacterial infections (NCT02714595). Patients with nosocomial pneumonia, bloodstream infections, sepsis, or cUTI were included in the trial. BAT regimens varied according to local practices and consisted of 1 to 3 antibacterial drugs with activity against Gram-negative bacteria. Most of the BAT regimens contained colistin.
The increase in all-cause mortality occurred in patients treated for nosocomial pneumonia, bloodstream infections, or sepsis. The 28-Day all-cause mortality was higher in patients treated with FETROJA than in patients treated with BAT [25/101 (24.8%) vs. 9/49 (18.4%), treatment difference 6.4%, 95% CI (-8.6, 19.2)]. All-cause mortality remained higher in patients treated with FETROJA than in patients treated with BAT through Day 49 [34/101 (33.7%) vs. 10/49 (20.4%), treatment difference 13.3%, 95% CI (-2.5, 26.9)]. Generally, deaths were in patients with infections caused by Gram-negative organisms, including non-fermenters such as Acinetobacter baumannii complex, Stenotrophomonas maltophilia , and Pseudomonas aeruginosa , and were the result of worsening or complications of infection, or underlying comorbidities. The cause of the increase in mortality has not been established.
Closely monitor the clinical response to therapy in patients with cUTI and HABP/VABP.
Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) reactions and serious skin reactions have been reported in patients receiving beta-lactam antibacterial drugs. Hypersensitivity was observed in FETROJA-treated patients in clinical trials [see Adverse Reactions (6.1) ] . These reactions are more likely to occur in individuals with a history of beta-lactam hypersensitivity and/or a history of sensitivity to multiple allergens. There have been reports of individuals with a history of penicillin hypersensitivity who have experienced severe reactions when treated with cephalosporins.
Before therapy with FETROJA is instituted, inquire about previous hypersensitivity reactions to cephalosporins, penicillins, or other beta-lactam antibacterial drugs. Discontinue FETROJA if an allergic reaction occurs.
Clostridioides difficile -associated Diarrhea (CDAD)
Clostridioides difficile -associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial agents, including FETROJA. CDAD may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of C . difficile .
C. difficile produces toxins A and B, which contribute to the development of CDAD. Hypertoxin-producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, antibacterial drugs not directed against C. difficile may need to be discontinued. Manage fluid and electrolyte levels as appropriate, supplement protein intake, monitor antibacterial treatment of C. difficile , and institute surgical evaluation as clinically indicated.
Seizures and Other Central Nervous System (CNS) Adverse Reactions
Cephalosporins, including FETROJA, have been implicated in triggering seizures [see Adverse Reactions (6.1) ] . Nonconvulsive status epilepticus (NCSE), encephalopathy, coma, asterixis, neuromuscular excitability, and myoclonia have been reported with cephalosporins particularly in patients with a history of epilepsy and/or when recommended dosages of cephalosporins were exceeded due to renal impairment. Adjust FETROJA dosing based on creatinine clearance [see Dosage and Administration (2.2) ] . Anticonvulsant therapy should be continued in patients with known seizure disorders. If CNS adverse reactions including seizures occur, patients should undergo a neurological evaluation to determine whether FETROJA should be discontinued.
Development of Drug-Resistant Bacteria
Prescribing FETROJA in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria [see Indications and Usage (1.3) ] .
ADVERSE REACTIONS
The following serious adverse reactions are described in greater detail in the Warnings and Precautions section:
- Increase in All-Cause Mortality in Patients with Carbapenem-Resistant Gram-Negative Bacterial Infections [see Warnings and Precautions (5.1) ]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2) ]
- Clostridioides difficile -associated Diarrhea (CDAD) [see Warnings and Precautions (5.3) ]
- Seizures and Other Central Nervous System Adverse Reactions [see Warnings and Precautions (5.4) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Complicated Urinary Tract Infections (cUTIs), Including Pyelonephritis
FETROJA was evaluated in an active-controlled, randomized clinical trial in patients with cUTI, including pyelonephritis (Trial 1). In this trial, 300 patients received FETROJA 2 grams every 8 hours infused over 1 hour (or a renally-adjusted dose), and 148 patients were treated with imipenem/cilastatin 1gram/1gram every 8 hours infused over 1 hour (or a renally-adjusted dose). The median age of treated patients across treatment arms was 65 years (range 18 to 93 years), with approximately 53% of patients aged greater than or equal to 65. Approximately 96% of patients were White, most were from Europe, and 55% were female. Patients across treatment arms received treatment for a median duration of 9 days.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In Trial 1, a total of 14/300 (4.7%) cUTI patients treated with FETROJA and 12/148 (8.1%) of cUTI patients treated with imipenem/cilastatin experienced serious adverse reactions. One death (0.3%) occurred in 300 patients treated with FETROJA as compared to none treated with imipenem/cilastatin. Discontinuation of treatment due to any adverse reaction occurred in 5/300 (1.7%) of patients treated with FETROJA and 3/148 (2.0%) of patients treated with imipenem/cilastatin. Specific adverse reactions leading to treatment discontinuation in patients who received FETROJA included diarrhea (0.3%), drug hypersensitivity (0.3%), and increased hepatic enzymes (0.3%).
Common Adverse Reactions
Table 4 lists the most common selected adverse reactions occurring in ≥ 2% of cUTI patients receiving FETROJA in Trial 1.
| Adverse Reaction | FETROJA 2 grams IV over 1 hour every 8 hours (with dosing adjustment based on renal function). (N = 300) | Imipenem/Cilastatin 1 gram IV over 1 hour every 8 hours (with dosing adjustment based on renal function and body weight). (N = 148) |
|---|---|---|
| cUTI = complicated urinary tract infection. | ||
| Diarrhea | 4% | 6% |
| Infusion site reactions Infusion site reactions include infusion site erythema, inflammation, pain, pruritus, injection site pain, and phlebitis. | 4% | 5% |
| Constipation | 3% | 4% |
| Rash Rash includes rash macular, rash maculopapular, erythema, skin irritation. | 3% | < 1% |
| Candidiasis Candidiasis includes oral or vulvovaginal candidiasis, candiduria. | 2% | 3% |
| Cough | 2% | < 1% |
| Elevations in liver tests Elevations in liver tests include alanine aminotransferase, aspartate aminotransferase, gamma-glutamyl transferase, blood alkaline phosphatase, hepatic enzyme increased. | 2% | < 1% |
| Headache | 2% | 5% |
| Hypokalemia Hypokalemia includes blood potassium decreased. | 2% | 3% |
| Nausea | 2% | 4% |
| Vomiting | 2% | 1% |
Other Adverse Reactions of FETROJA in the cUTI Patients (Trial 1)
The following selected adverse reactions were reported in FETROJA-treated cUTI patients at a rate of less than 2% in Trial 1:
Blood and lymphatic disorders : thrombocytosis
Cardiac disorders : congestive heart failure, bradycardia, atrial fibrillation
Gastrointestinal disorders : abdominal pain, dry mouth, stomatitis
General system disorders : pyrexia, peripheral edema
Hepatobiliary disorders : cholelithiasis, cholecystitis, gallbladder pain
Immune system disorders : drug hypersensitivity
Infections and infestations : C. difficile infection
Laboratory investigations : prolonged prothrombin time (PT) and prothrombin time international normalized ratio (PT-INR), red blood cells urine positive, creatine phosphokinase increase
Metabolism and nutrition disorders : decreased appetite, hypocalcemia, fluid overload
Nervous system disorders : dysgeusia, seizure
Respiratory, thoracic, and mediastinal disorders : dyspnea, pleural effusion
Skin and subcutaneous tissue disorders : pruritus
Psychiatric disorders : insomnia, restlessness
Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
FETROJA was evaluated in an active-controlled clinical trial in patients with HABP/VABP (Trial 2). In this trial, 148 patients received FETROJA 2 grams every 8 hours infused over 3 hours, and 150 patients received meropenem 2 grams every 8 hours infused over 3 hours. Doses of study treatments were adjusted based on renal function. The median age was 67 years, approximately 59% of patients were 65 years of age and older, 69% were male, and 68% were White. Overall, approximately 60% were ventilated at randomization, including 41% with VABP and 14% with ventilated HABP. The mean Acute Physiology And Chronic Health Evaluation (APACHE II) score was 16. All patients received empiric treatment for Gram-positive organisms with linezolid for at least 5 days.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In Trial 2, serious adverse reactions occurred in 54/148 (36.5%) HABP/VABP patients treated with FETROJA and 45/150 (30%) of HABP/VABP patients treated with meropenem. Adverse reactions leading to death were reported in 39/148 (26.4%) patients treated with FETROJA and 35/150 (23.3%) patients treated with meropenem. Adverse reactions leading to discontinuation of treatment occurred in 12/148 (8.1%) of patients treated with FETROJA and 14/150 (9.3%) of patients treated with meropenem. The most common adverse reactions leading to discontinuation in both treatment groups were elevated liver tests.
Common Adverse Reactions
Table 5 lists the most common selected adverse reactions occurring in ≥ 4% of patients receiving FETROJA in the HABP/VABP trial.
| Adverse Reaction | FETROJA 2 grams IV over 3 hours every 8 hours (with dosing adjustment based on renal function). N = 148 | Meropenem 2 grams IV over 3 hours every 8 hours (with dosing adjustment based on renal function). N = 150 |
|---|---|---|
| HABP/VABP = hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia. | ||
| Elevations in liver tests Elevations in liver tests include the following terms: aspartate aminotransferase increased, alanine aminotransferase increased, gamma-glutamyl transferase increased, liver function test increased, liver function test abnormal, hepatic enzyme increased, transaminases increased, hypertransaminesemia. | 16% | 16% |
| Hypokalemia Hypokalemia includes blood potassium decreased. | 11% | 15% |
| Diarrhea | 9% | 9% |
| Hypomagnesemia | 5% | < 1% |
| Atrial fibrillation | 5% | 3% |
Other Adverse Reactions of FETROJA in HABP/VABP Patients in Trial 2
The following selected adverse reactions were reported in FETROJA-treated HABP/VABP patients at a rate of less than 4% in Trial 2:
Blood and lymphatic disorders : thrombocytopenia, thrombocytosis
Cardiac disorders : myocardial infarction, atrial flutter
Gastrointestinal disorders : nausea, vomiting, abdominal pain
Hepatobiliary disorders : cholecystitis, cholestasis
Infections and infestations : C. difficile infection, oral candidiasis
Laboratory investigations : prolonged prothrombin time (PT) and prothrombin time international normalized ratio (PT-INR), activated partial thromboplastin time (aPTT)
Metabolism and nutrition disorders : hypocalcemia, hyperkalemia
Nervous system disorders : seizure
Renal and genitourinary disorders : acute interstitial nephritis
Respiratory, thoracic, and mediastinal disorders : cough
Skin and subcutaneous tissue disorders : rash including rash erythematous
Postmarketing Experience
The following adverse reactions have been identified during postapproval use of FETROJA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders : neutropenia, eosinophilia
Renal and urinary disorders : chromaturia
DRUG INTERACTIONS
Use alternate testing methods to confirm positive results of dipstick tests (urine protein, ketones, or occult blood). (7.1 )
Drug/Laboratory Test Interactions
Cefiderocol may result in false-positive results in dipstick tests (urine protein, ketones, or occult blood). Use alternate clinical laboratory methods of testing to confirm positive tests.
DESCRIPTION
FETROJA is a cephalosporin antibacterial drug product consisting of cefiderocol sulfate tosylate for intravenous infusion. Cefiderocol functions as a siderophore [see Microbiology (12.4) ] .
The chemical name of cefiderocol sulfate tosylate is Tris[(6 R ,7 R )-7-[(2 Z )-2-(2-amino-1,3-thiazol-4-yl)-2-{[(2-carboxypropan-2-yl)oxy]imino}acetamido]-3-({1-[2-(2-chloro-3,4-dihydroxybenzamido)ethyl]pyrrolidin-1-ium-1-yl}methyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate] tetrakis(4-methylbenzenesulfonate) monosulfate hydrate, and the molecular weight is 3043.50 (anhydrous). The molecular formula is 3C 30 H 34 ClN 7 O 10 S 2 ∙4C 7 H 8 O 3 S∙H 2 SO 4 ∙xH 2 O.
| Figure 1 Chemical Structure of Cefiderocol Sulfate Tosylate |
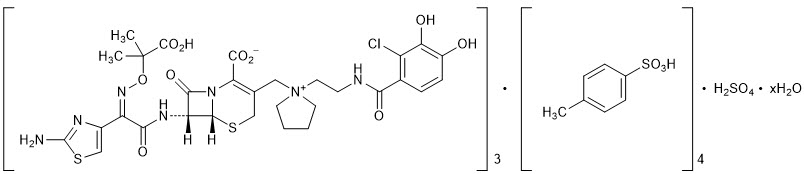 |
FETROJA for injection is a white to off-white, sterile, lyophilized powder formulated with 1 gram of cefiderocol (equivalent to 1.6 grams of cefiderocol sulfate tosylate), sucrose (900 mg), sodium chloride (216 mg), and sodium hydroxide to adjust pH. The sodium content is approximately 176 mg/vial. The pH of the reconstituted solution of 1 gram cefiderocol (1 vial) dissolved in 10 mL water is 5.2 to 5.8.
CLINICAL PHARMACOLOGY
Mechanism of Action
FETROJA is an antibacterial drug [see Microbiology (12.4) ] .
Pharmacodynamics
The percent time of dosing interval that unbound plasma concentrations of cefiderocol exceed the minimum inhibitory concentration (MIC) against the infecting organism best correlates with antibacterial activity in neutropenic murine thigh and lung infection models with E. coli , K. pneumoniae , P. aeruginosa , A. baumannii , and S. maltophilia . Compared to a 1-hour infusion, a 3-hour infusion increased the percent time of dosing interval that unbound plasma concentrations of cefiderocol exceed the MIC. The in vivo animal pneumonia studies showed that the antibacterial activity of cefiderocol was greater at the human equivalent dosing regimen of 3-hour infusion compared to that of 1-hour infusion.
Cardiac Electrophysiology
At doses 1 and 2 times the maximum recommended dosage, FETROJA does not prolong the QT interval to any clinically relevant extent.
Pharmacokinetics
Cefiderocol exposures (C max and daily AUC) in cUTI patients, HABP/VABP patients, and healthy volunteers are summarized in Table 6. Cefiderocol C max and AUC increased proportionally with dose.
| PK Parameters | cUTI Patients After multiple (every 8 hours) FETROJA 2-gram doses infused over 3 hours or adjusted based on renal function. (N = 21) | HABP/VABP Patients (N = 146) | Healthy Volunteers After a single FETROJA 2-gram dose was infused over 3 hours. (N = 43) |
|---|---|---|---|
| C max = maximum concentration. | |||
| AUC 0-24 hr = area under the concentration time curve from 0 to 24 hours. | |||
| C max (mg/L) | 115 (±57) | 111 (±56) | 91.4 (±17.9) |
| AUC 0-24 hr (mg∙hr/L) | 1944 (±1097) | 1773 (±990) | 1175 (±203) |
Distribution
The geometric mean (±SD) cefiderocol volume of distribution was 18.0 (±3.36) L. Plasma protein binding, primarily to albumin, of cefiderocol is 40% to 60%.
Following a FETROJA 2-gram dose (or renal function equivalent dose) at steady state in patients with pneumonia requiring mechanical ventilation with a 3-hour infusion, the cefiderocol concentrations in epithelial lining fluid ranged from 3.1 to 20.7 mg/L and 7.2 to 15.9 mg/L at the end of infusion and at 2 hours after the end of infusion, respectively.
Elimination
Cefiderocol terminal elimination half-life is 2 to 3 hours. The geometric mean (±SD) cefiderocol clearance is estimated to be 5.18 (±0.89) L/hr.
Metabolism
Cefiderocol is minimally metabolized [less than 10% of a single radiolabeled cefiderocol dose of 1 gram (0.5 times the approved recommended dosage) infused over 1 hour].
Excretion
Cefiderocol is primarily excreted by the kidneys. After a single radiolabeled cefiderocol 1-gram (0.5 times the approved recommended dosage) dose infused over 1 hour, 98.6% of the total radioactivity was excreted in urine (90.6% unchanged) and 2.8% in feces.
Specific Populations
No clinically significant differences in the pharmacokinetics of cefiderocol were observed based on age (18 to 93 years of age), sex, or race. The effect of hepatic impairment on the pharmacokinetics of cefiderocol was not evaluated.
Patients with Renal Impairment
Approximately 60% of cefiderocol was removed by a 3- to 4-hour hemodialysis session.
Cefiderocol AUC fold changes in subjects with renal impairment compared to subjects with CLcr 90 to 119 mL/min are summarized in Table 7.
| CLcr (mL/min) | Cefiderocol AUC Geometric Mean Ratios (90% CI) Compared to AUC in subjects with CLcr 90 to 119 mL/min (N = 12). |
|---|---|
| CI = confidence interval. | |
| 60 to 89 (N = 6) | 1.37 (1.15, 1.62) |
| 30 to 59 (N = 7) | 2.35 (2.00, 2.77) |
| 15 to 29 (N = 4) | 3.21 (2.64, 3.91) |
| < 15 (N = 6) | 4.69 (3.95, 5.56) |
Patients Receiving CRRT
In an in vitro study, effluent flow rate was the major determinant of cefiderocol clearance by CRRT. Variables examined included effluent flow rate, CRRT mode (CVVH or CVVHD), filter type and point of dilution (pre- or post-filter dilution). The effluent flow rate-based dosing recommendations in Table 2 are predicted to provide cefiderocol exposures similar to those achieved with a dose of 2 grams given every 8 hours in patients not receiving CRRT [see Dosage and Administration (2.2) ] .
Patients with CLcr 120 mL/min or Greater
Increased cefiderocol clearance has been observed in patients with CLcr 120 mL/min or greater. A FETROJA 2-gram dose every 6 hours infused over 3 hours provided cefiderocol exposures comparable to those in patients with CLcr 90 to 119 mL/min [see Dosage and Administration (2.2) ] .
Drug Interaction Studies
Clinical Studies
No clinically significant differences in the pharmacokinetics of furosemide (an organic anion transporter [OAT]1 and OAT3 substrate), metformin (an organic cation transporter [OCT]1, OCT2, and multidrug and toxin extrusion [MATE]2-K substrate), and rosuvastatin (an organic anion transporting polypeptide [OATP]1B3 substrate) were observed when coadministered with cefiderocol.
In Vitro Studies Where Drug Interaction Potential Was Not Further Evaluated Clinically
Cytochrome P450 (CYP) Enzymes : Cefiderocol is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4. Cefiderocol is not an inducer of CYP1A2, CYP2B6, or CYP3A4.
Transporter Systems : Cefiderocol is not an inhibitor of OATP1B1, MATE1, P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), or bile salt export pump transporters. Cefiderocol is not a substrate of OAT1, OAT3, OCT2, MATE1, MATE2-K, P-gp, or BCRP.
Microbiology
Mechanism of Action
FETROJA is a cephalosporin antibacterial with activity against Gram-negative aerobic bacteria. Cefiderocol functions as a siderophore and binds to extracellular free (ferric) iron. In addition to passive diffusion via porin channels, cefiderocol is actively transported across the outer cell membrane of bacteria into the periplasmic space using the bacterial siderophore iron uptake mechanism. Cefiderocol exerts bactericidal action by inhibiting cell wall biosynthesis through binding to penicillin-binding proteins (PBPs).
Cefiderocol has no clinically relevant in vitro activity against most Gram-positive bacteria and anaerobic bacteria.
Resistance
In vitro, MIC increases that may result in resistance to cefiderocol in Gram-negative bacteria have been associated with a combination of multiple beta-lactamases, modifications of PBPs, and mutations of transcriptional regulators that impact siderophore expression.
Cefiderocol does not cause induction of AmpC beta-lactamase in P. aeruginosa and E. cloacae . The frequency of resistance development in Gram-negative bacteria including carbapenemase producers exposed to cefiderocol at 10× minimum inhibitory concentration (MIC) ranged from 10 -6 to < 10 -8 .
Cross-resistance with other classes of antibacterial drugs has not been identified; therefore, isolates resistant to other antibacterial drugs may be susceptible to cefiderocol.
Cefiderocol has shown in vitro activity against isolates of S. maltophilia and a subset of isolates of Enterobacterales and P. aeruginosa that are resistant to meropenem, ciprofloxacin, amikacin, cefepime, ceftazidime-avibactam, and ceftolozane/tazobactam . Cefiderocol has shown in vitro activity against subset of isolates of A. baumannii complex that are resistant to meropenem, ciprofloxacin, and amikacin . Cefiderocol is active against some colistin-resistant E. coli isolates containing mcr-1 .
Cefiderocol demonstrated in vitro activity against a subgroup of Enterobacterales genetically confirmed to contain the following: ESBLs (TEM, SHV, CTX-M, oxacillinase [OXA]), AmpC, AmpC-type ESBL (CMY), serine-carbapenemases (such as KPC, OXA-48), and metallo-carbapenemases (such as NDM and VIM). Cefiderocol demonstrated in vitro activity against a subgroup of P. aeruginosa genetically confirmed to contain VIM, IMP, GES, AmpC, and a subgroup of A. baumannii containing OXA-23, OXA-24/40, OXA-51, OXA-58, and AmpC. Cefiderocol is active in vitro against a subgroup of S. maltophilia containing metallo-carbapenemase (L1) and serine beta-lactamases (L2).
Cefiderocol maintained in vitro activity against K. pneumoniae in the presence of porin channel deletions (OmpK35/36), and against P. aeruginosa in the presence of porin channel deletions (OprD) and efflux pump up-regulation (MexAB-OprM, MexCD-OprJ, MexEF-OprN, and MexXY).
In vitro, the addition of the beta-lactamase inhibitors (such as avibactam, clavulanic acid, and dipicolinic acid) results in the lowering of MICs of some clinical isolates with relatively high MICs (range 2 to 256 mcg/mL) to cefiderocol.
Interaction with Other Antimicrobials
In vitro studies showed no antagonism between cefiderocol and amikacin, ceftazidime/avibactam, ceftolozane/tazobactam, ciprofloxacin, clindamycin, colistin, daptomycin, linezolid, meropenem, metronidazole, tigecycline, or vancomycin against strains of Enterobacterales, P. aeruginosa , and A. baumannii .
Activity against Bacteria in Animal Infection Models
In a neutropenic murine thigh infection model using a humanized dose (2 grams every 8 hours), cefiderocol demonstrated 1log 10 reduction in bacterial burden against most E. coli, K. pneumoniae , A. baumannii , S. maltophilia, and P. aeruginosa including some carbapenemase-producing (KPC, OXA-23, OXA-24/40, OXA-58) isolates with MICs of ≤ 4 mcg/mL to cefiderocol.
In an immunocompetent rat pneumonia model, reduction in bacterial counts in the lungs of animals infected with K. pneumoniae with MICs ≤ 8 mcg/mL, A. baumannii with MICs ≤ 2 mcg/mL , and P. aeruginosa with MICs ≤ 1 mcg/mL including carbapenemase-producing (KPC, NDM, and IMP) isolates was observed using humanized cefiderocol drug exposure.
In an immunocompetent murine urinary tract infection model, cefiderocol reduced bacterial counts in the kidneys of mice infected with E. coli, K. pneumoniae , and P. aeruginosa isolates with MICs ≤ 1 mcg/mL. In an immunocompromised murine systemic infection model, cefiderocol increased survival in mice infected with E. cloacae , S. maltophilia , and Burkholderia cepacia isolates with MICs ≤ 0.5 mcg/mL compared to untreated mice. In an immunocompetent murine systemic infection model, cefiderocol increased survival in mice infected with S. marcescens and P. aeruginosa isolates with MICs ≤ 1 mcg/mL compared to untreated mice.
The clinical significance of the above findings in animal infection models is not known.
Antimicrobial Activity
FETROJA has been shown to be active against the following bacteria, both in vitro and in clinical infections [see Indications and Usage (1) ] :
Complicated Urinary Tract Infections, Including Pyelonephritis
Gram-negative Bacteria
Escherichia coli
Enterobacter cloacae complex
Klebsiella pneumoniae
Proteus mirabilis
Pseudomonas aeruginosa
Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
Gram-negative Bacteria
Acinetobacter baumannii complex
Escherichia coli
Enterobacter cloacae complex
Klebsiella pneumoniae
Pseudomonas aeruginosa
Serratia marcescens
The following in vitro data are available, but their clinical significance is not known. At least 90% of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for FETROJA against isolates of similar genus or organism group. However, the efficacy of FETROJA in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-negative Bacteria
Achromobacter spp.
Burkholderia cepacia complex
Citrobacter freundii complex
Citrobacter koseri
Klebsiella aerogenes
Klebsiella oxytoca
Morganella morganii
Proteus vulgaris
Providencia rettgeri
Stenotrophomonas maltophilia
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see https://www.fda.gov/STIC.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies in animals have not been conducted with cefiderocol.
Mutagenesis
Cefiderocol was negative for genotoxicity in a reverse mutation test with S. typhimurium and E. coli and did not induce mutations in V79 Chinese hamster lung cells. Cefiderocol was positive in a chromosomal aberration test in cultured TK6 human lymphoblasts and increased mutation frequency in L5178Y mouse lymphoma cells. Cefiderocol was negative in an in vivo rat micronucleus test and a rat comet assay at the highest doses of 2000 and 1500 mg/kg/day, respectively.
Impairment of Fertility
Cefiderocol did not affect fertility in adult male or female rats when administered intravenously at doses up to 1000 mg/kg/day. The AUC at this dose is approximately 0.9 times the mean daily cefiderocol exposure in patients who received the maximum recommended clinical dose of 2 grams every 8 hours.
CLINICAL STUDIES
Complicated Urinary Tract Infections, Including Pyelonephritis
A total of 448 adults hospitalized with cUTI (including pyelonephritis) were randomized in a 2:1 ratio and received study medications in a multinational, double-blind trial (Trial 1) (NCT02321800) comparing FETROJA 2 grams intravenously (IV) every 8 hours (infused over 1 hour) to imipenem/cilastatin 1gram/1gram IV every 8 hours (infused over 1 hour) for 7 to 14 days. No switch from IV to oral antibacterial therapy was permitted.
Efficacy was assessed as a composite of microbiological eradication and clinical cure at the Test-of-Cure visit (TOC) in the microbiological intent-to-treat (Micro-ITT) population, which included all patients who received at least a single dose of study medication and had at least one baseline Gram-negative uropathogen. Other efficacy endpoints included the microbiological eradication rate and the clinical response rate at TOC in the Micro-ITT population.
The Micro-ITT population consisted of 371 patients of whom 25% had cUTI with pyelonephritis, 48% had cUTI without pyelonephritis, and 27% had acute uncomplicated pyelonephritis. Complicating conditions included obstructive uropathy, catheterization, and renal stones. The median age was 66 years, with 24% of patients over the age of 75 years, and 55% of the population were female. The median duration of therapy in both treatment groups was 9 days (range 1-14 days). Of the 371 patients, 32% had CLcr > 50-80 mL/min, 17% had CLcr 30-50 mL/min, and 3% had CLcr < 30 mL/min at baseline. Concomitant Gram-negative bacteremia was identified in 7% of patients. In the Micro-ITT population, the most common baseline pathogens were E. coli and K. pneumoniae .
Table 8 provides the results of a composite of microbiological eradication (all Gram-negative uropathogens found at baseline at ≥ 10 5 CFU/mL reduced to < 10 4 CFU/mL) and clinical response (resolution or improvement of cUTI symptoms and no new symptoms assessed by the investigator) at the TOC visit, 7+/-2 days after the last dose of study drug. The response rates for the composite endpoint of microbiological eradication and clinical response at the TOC visit were higher in the FETROJA arm compared with imipenem/cilastatin, as shown in Table 8. Clinical response rates at the TOC visit were similar between FETROJA and imipenem/cilastatin. Most patients with microbiological failure at the TOC visit in either treatment arm did not require further antibacterial drug treatment. Subgroup analyses examining composite outcomes by baseline pathogen are shown in Table 9 and demonstrated responses consistent with the overall population. Subgroup analyses examining outcomes by age, gender, and/or outcomes in patients with renal impairment, concomitant bacteremia, complicated UTI with or without pyelonephritis, or acute uncomplicated pyelonephritis demonstrated responses were consistent with the overall population.
| Study Endpoint | FETROJA n/N (%) | Imipenem/Cilastatin n/N (%) | Treatment Difference (95% CI) The treatment difference and 95% CI were based on the Cochran-Mantel-Haenszel method. |
|---|---|---|---|
| CI = confidence interval; Micro-ITT = microbiological intent-to-treat; TOC = Test of Cure. | |||
| Composite response at TOC | 183/252 (72.6%) | 65/119 (54.6%) | 18.6 (8.2, 28.9) |
| Microbiologic response TOC | 184/252 (73.0%) | 67/119 (56.3%) | 17.3 (6.9, 27.6) |
| Clinical response TOC | 226/252 (89.7%) | 104/119 (87.4%) | 2.4 (-4.7, 9.4) |
| Baseline Pathogen Subgroup | FETROJA n/N (%) | Imipenem/Cilastatin n/N (%) |
|---|---|---|
| Escherichia coli | 113/152 (74.3) | 45/79 (57.0) |
| Klebsiella pneumoniae | 36/48 (75.0) | 12/25 (48.0) |
| Proteus mirabilis | 13/17 (76.5) | 0/2 (0.0) |
| Pseudomonas aeruginosa | 8/18 (44.4) | 3/5 (60.0) |
| Enterobacter cloacae complex | 8/13 (61.5) | 3/3 (100.0) |
In the FETROJA treatment group, 61 (24.2%) bacterial isolates were ESBL producers compared with 32 (26.9%) in the imipenem/cilastatin group. The composite response rate of patients with these ESBL isolates at the TOC visit was consistent with the overall results.
Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
A total of 298 hospitalized adults with HABP/VABP received study medications in a multicenter, randomized, double-blind trial (Trial 2) (NCT03032380) comparing FETROJA 2 grams administered intravenously every 8 hours as a 3-hour infusion to meropenem (2 grams every 8 hours infused over 3 hours). Dosing was adjusted for renal function. Patients in both treatment arms received linezolid 600 mg every 12 hours for at least 5 days for empiric treatment of Gram-positive organisms. The trial protocol permitted administration of potentially active prior antibacterial therapy for no more than 24 hours within 72 hours prior to randomization and disallowed systemic concomitant antibacterial therapy until the Test-of-Cure visit (TOC, 7 days after end of treatment). The analysis population was the modified intent-to-treat (mITT) population, which included all randomized patients who received study medication and had evidence of bacterial pneumonia, except those with only anaerobic or Gram-positive aerobic infections.
Of the 292 patients in the mITT population, the median age was 67 years, and 58% of the population was 65 years of age and older, with 29% of the population 75 years of age and older. The majority of patients were male (68%), White (69%), and were from Europe (67%). Approximately 4% (11/292) were from the United States. The median baseline APACHE II score was 15, and 29% of patients had a baseline APACHE II score of greater than or equal to 20. At randomization, 68% of patients were in the ICU, and 60% were mechanically ventilated. 60% of patients had CLcr less than or equal to 80 mL/min at baseline; among these, 34% had CLcr less than or equal to 50 mL/min, and 14% had a CLcr less than 30 mL/min. Augmented renal clearance (CLcr greater than 120 mL/min) was present in 16% of patients. Gram-negative bacteremia was present at baseline in 6% of patients. In both treatment groups, most patients (70%) received between 7 and 14 days of study medication and 18% between 15 and 21 days.
Table 10 shows the Day 14 and Day 28 all-cause mortality rates, as well as clinical cure at the TOC visit. FETROJA was noninferior to meropenem with regard to the primary efficacy endpoint (Day 14 all-cause mortality in the mITT population). Clinical cure was defined as resolution or substantial improvement in signs and symptoms associated with pneumonia, such that no additional antibacterial therapy was required for the treatment of the current infection through the TOC visit.
| Endpoint | FETROJA n/N (%) | Meropenem n/N (%) | Treatment Difference The adjusted treatment difference (FETROJA minus meropenem) and associated 95% CI were based on the Cochran-Mantel-Haenszel stratum-weighted method. Subjects with unknown survival status were considered deaths. For Day 14 All-cause Mortality, 1 meropenem subject had unknown status; for Day 28 All-cause Mortality, 1 meropenem subject and 2 FETROJA subjects had unknown status. (95% CI) |
|---|---|---|---|
| CI = confidence interval; TOC = Test of Cure. | |||
| Day 14 All-cause Mortality | 18/145 (12.4) | 18/147 (12.2) | 0.2 (-7.2, 7.7) |
| Day 28 All-cause Mortality | 32/145 (22.1) | 31/147 (21.1) | 1.1 (-8.2, 10.4) |
| Clinical Cure at TOC | 94/145 (64.8) | 98/147 (66.7) | -2.0 (-12.5, 8.5) |
The Day 14 and Day 28 all-cause mortality rates by pathogen in patients in the mITT population who had a baseline LRT pathogen that was susceptible to meropenem are shown in Table 11; the clinical outcome at the TOC visit is shown in Table 12. There were 51 patients with A. baumannii complex at baseline, of which 17 (33.3%) patients had isolates susceptible to meropenem (MIC ≤ 8 mcg/mL, based on meropenem 2 grams every 8 hours). Among 51 patients with A. baumannii complex at baseline, all-cause mortality at Day 14 was 5/26 (19.2%) in FETROJA and 4/25 (16.0%) in the meropenem treatment group and at Day 28 was 9/26 (34.6%) in FETROJA and 6/25 (24.0%) in the meropenem treatment group. The clinical cure rates at the TOC visit were 14/26 (53.8%) in the FETROJA and 15/25 (60.0%) in the meropenem treatment group.
| Baseline Pathogen | Day 14 All-cause Mortality | Day 28 All-cause Mortality | ||
|---|---|---|---|---|
| FETROJA n/N (%) | Meropenem n/N (%) | FETROJA n/N (%) | Meropenem n/N (%) | |
| Each cell excludes subjects in whom baseline pathogen had meropenem MIC > 8 mcg/mL or where MIC was unknown. Subjects with unknown survival status were considered deaths. | ||||
| Klebsiella pneumoniae | 4/38 (10.5) | 4/36 (11.1) | 8/38 (21.1) | 9/36 (25.0) |
| Pseudomonas aeruginosa | 2/20 (10.0) | 4/17 (23.5) | 2/20 (10.0) | 5/17 (29.4) |
| Acinetobacter baumannii complex Includes A. baumannii, A. nosocomialis, and A. pittii. | 1/8 (12.5) | 0/9 (0.0) | 3/8 (37.5) | 0/9 (0.0) |
| Escherichia coli | 3/18 (16.7) | 3/21 (14.3) | 5/18 (27.8) | 4/21 (19.0) |
| Other Enterobacterales Includes Enterobacter cloacae complex ( E. cloacae, E. asburiae, and E. kobei) and Serratia marcescens. | 2/16 (12.5) | 2/14 (14.3) | 4/16 (25.0) | 3/14 (21.4) |
| Baseline Pathogen | Clinical Cure | |
|---|---|---|
| FETROJA n/N (%) | Meropenem n/N (%) | |
| Each cell excludes subjects whose pathogen-specific meropenem MIC was > 8 mcg/mL or where MIC was unknown. | ||
| Klebsiella pneumoniae | 24/38 (63.2) | 23/36 (63.9) |
| Pseudomonas aeruginosa | 13/20 (65.0) | 13/17 (76.5) |
| Acinetobacter baumannii complex Includes A. baumannii, A. nosocomialis, and A. pittii. | 6/8 (75.0) | 7/9 (77.8) |
| Escherichia coli | 12/18 (66.7) | 13/21 (61.9) |
| Other Enterobacterales Includes Enterobacter cloacae complex ( E. cloacae, E. asburiae, and E. kobei) and Serratia marcescens. | 10/16 (62.5) | 8/14 (57.1) |
In the FETROJA treatment group, 45 (31%) patients had ESBL-producing bacterial isolates compared with 42 (28.6%) patients in the meropenem treatment group. All-cause mortality at Day 14 and Day 28 of patients with these ESBL-producing bacterial isolates was consistent with the overall results.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
FETROJA 1 gram (cefiderocol) for injection is supplied as a white to off-white sterile lyophilized powder for reconstitution in single-dose, clear glass vials (NDC 59630-266-01) sealed with a rubber stopper (not made with natural rubber latex) and an aluminum seal with flip-off cap. Each vial is supplied in cartons containing 10 single-dose vials.
| NDC 59630-266-10 | FETROJA (cefiderocol) 1 gram/vial, 10 vials/carton |
Storage and Handling
FETROJA vials should be stored refrigerated at 2°C to 8°C (36°F to 46°F). Protect from light. Store in the carton until time of use. Store reconstituted solutions of FETROJA at room temperature [see Dosage and Administration (2.5) ] .
Mechanism of Action
FETROJA is an antibacterial drug [see Microbiology (12.4) ] .