Get your patient on Fetzima (Levomilnacipran Hydrochloride)
Dosage & administration

Fetzima prescribing information
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors [seeWarnings and Precautions (5.1 )] .
FETZIMA is not approved for use in pediatric patients [seeUse in Specific Populations (8.4 )] .
1 INDICATIONS AND USAGE
FETZIMA ® is indicated for the treatment of major depressive disorder (MDD) in adults [see Clinical Studies (14 )].
Limitation of Use: FETZIMA is not approved for the management of fibromyalgia. The efficacy and safety of FETZIMA for the management of fibromyalgia have not been established.
2 DOSAGE AND ADMINISTRATION
- Recommended dosage: 40 mg to 120 mg once daily with or without food (2.1 ).
- Initial dosage is 20 mg once daily for 2 days and then increase to 40 mg once daily (2.1 ).
- Based on clinical response and tolerability, increase dose in increments of 40 mg at intervals of 2 or more days (2.1 ).
- The maximum recommended dosage is 120 mg once daily (2.1 ).
- Take capsules whole; do not open, chew or crush (2.1 )
- Renal impairment (2.3 ) : ○ Severe renal impairment: Maximum recommended dosage is 40 mg once daily. ○ Moderate renal impairment: Maximum recommended dosage is 80 mg once daily.
- Discontinuation : Reduce dose gradually whenever possible (2.4 )
2.1 Recommended Dosage
The recommended dosage range for FETZIMA is 40 mg to 120 mg once daily, with or without food. FETZIMA should be initiated at 20 mg once daily for 2 days and then increased to 40 mg once daily. Based on clinical response and tolerability, FETZIMA may be increased in increments of 40 mg at intervals of 2 or more days. The maximum recommended dosage is 120 mg once daily.
Take FETZIMA at approximately the same time each day. Swallow FETZIMA whole; do not open, chew, or crush the capsule.
2.2 Screen for Bipolar Disorder Prior to Starting FETZIMA
Prior to initiating treatment with FETZIMA or another antidepressant, screen patients for a personal or family history of bipolar disorder, mania, or hypomania [see Warnings and Precautions (5.8 ) ] .
2.000000000000000e+00 3 Dosage Recommendations for Patients with Renal Impairment
- End stage renal disease (ESRD): FETZIMA is not recommended;
- Severe renal impairment (creatinine clearance of 15 to 29 mL/min), the dosage should not exceed 40 mg once daily;
- Moderate renal impairment (creatinine clearance of 30 to 59 mL/min), the dosage should not exceed 80 mg once daily;
- Mild renal impairment (creatinine clearance of 60 to 89 mL/min): no dosage adjustment recommended [see Use in Specific Populations (8.7 )].
2.4 Discontinuing Treatment with FETZIMA
Discontinuation symptoms have been reported with discontinuation of serotonergic drugs such as FETZIMA. Gradual dose reduction is recommended, instead of abrupt discontinuation, whenever possible. Monitor patients for these symptoms when discontinuing FETZIMA. If intolerable symptoms occur following a dose decrease or upon discontinuation of treatment, consider resuming the previously prescribed dose and decreasing the dose at a more gradual rate [see Warnings and Precautions (5.10 )] .
2.5 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders
At least 14 days must elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with FETZIMA. Conversely, at least 7 days should be allowed after stopping FETZIMA before starting an MAOI antidepressant [see Contraindications (4 )] .
2.6 Dosage Recommendations for Use with Strong CYP3A4 Inhibitors
The maximum recommended dosage of FETZIMA should not exceed 80 mg once daily when used with strong CYP3A4 inhibitors [ s ee Drug Interactions (7.1 )] .
3 DOSAGE FORMS AND STRENGTHS
FETZIMA (levomilnacipran) is available as 20 mg, 40 mg, 80 mg, and 120 mg extended-release capsules.
| Capsule Strength | Capsule Color/Shape | Capsule Markings |
| 20 mg | yellow cap white body | black "FL" on cap black "20" on body |
| 40 mg | yellow cap yellow body | black "FL" on cap black "40" on body |
| 80 mg | pink cap white body | black "FL" on cap black "80" on body |
| 120 mg | pink cap pink body | black "FL" on cap black "120" on body |
8 USE IN SPECIFIC POPULATIONS
- Pregnancy : Third trimester use may increase risk for symptoms of poor adaptation (respiratory distress, temperature instability, feeding difficulty, hypotonia, tremor, irritability) in the neonate. (8.1 )
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antidepressants during pregnancy. Healthcare providers are encouraged to advise patients to register by calling the National Pregnancy Registry for Antidepressants at 1-844-405-6185 or visiting online at https://womensmentalhealth.org/research/pregnancyregistry/antidepressants .
Risk Summary
Based on data from published observational studies, exposure to SNRIs, particularly in the month before delivery, has been associated with a less than 2-fold increase in the risk of postpartum hemorrhage [see Warnings and Precautions (5.5 ) and Clinical Considerations] .
The available data on FETZIMA use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are risks associated with untreated depression in pregnancy and with exposure to SNRIs and SSRIs, including FETZIMA, during pregnancy (see Clinical Considerations ).
In animal reproduction studies, levomilnacipran was not associated with malformations in rats or rabbits when given during the period of organogenesis at doses up to 8 or 16 times the maximum recommended human dose (MRHD) of 120 mg on a mg/m 2 basis, respectively. However, an increase in early post-natal rat pup mortality was seen at a dose equivalent to 5 times the MRHD given during pregnancy and lactation (see Data ) .
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Women who discontinued antidepressants during pregnancy were more likely to experience a relapse of major depression than women who continued antidepressants. This finding is from a prospective, longitudinal study that followed 201 pregnant women with a history of major depressive disorder who were euthymic and taking antidepressants at the beginning of pregnancy. Consider the risk of untreated depression when discontinuing or changing treatment with antidepressant medication during pregnancy and postpartum.
Maternal Adverse Reactions
Use of FETZIMA in the month before delivery may be associated with an increased risk of postpartum hemorrhage [see Warnings and Precautions (5.5 )] .
Fetal/Neonatal adverse reactions
Neonates exposed to SNRIs or SSRIs, including FETZIMA, late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremor, jitteriness, irritability, and constant crying. These findings are consistent with either direct toxic effect of SSRIs and SNRIs or possibly, a drug discontinuation syndrome. It should be noted that, in some cases, the clinical picture is consistent with serotonin syndrome [see Warnings and Precautions (5.2 and 5.10 )].
Data
Animal Data
No malformations were observed when levomilnacipran was administered to pregnant rats or rabbits during the period of organogenesis at oral doses up to 100 mg/kg/day. This dose is 8 and 16 times (in rats and rabbits, respectively) the maximum recommended human dose (MRHD) of 120 mg on a mg/m 2 basis. Fetal body weights were reduced in rats, and skeletal ossification was delayed in both rats and rabbits at this dose; these effects were not observed in either species at doses up to 30 mg/kg/day, 2.4 times the MRHD in rats or 5 times the MRHD in rabbits on a mg/m 2 basis.
When levomilnacipran was administered to pregnant rats at an oral dose of 60 mg/kg/day, 5 times the MRHD, during organogenesis and throughout pregnancy and lactation, there was an increase in early postnatal pup mortality; no pup mortality was seen at 20 mg/kg/day, 1.6 times the MRHD on a mg/m 2 basis. Among the surviving pups, pre- and post-weaning pup weight gain was reduced up to at least 8 weeks of age; however, physical and functional development, including reproductive performance of the progeny, was not affected. The effects on body weight gain were not seen at 7 mg/kg/day, 0.6 times the MRHD on a mg/m 2 basis.
8.2 Lactation
Risk Summary
There are no available data on the presence of levomilnacipran in human milk; however, racemic milnacipran is present in human milk (see Data ). There are no reports on the effects of levomilnacipran or milnacipran on the breastfed infant or the effects on milk production. However, there are reports of agitation, irritability, poor feeding and poor weight gain in infants exposed to SSRIs or SNRIs through breast milk ( see Clinical Considerations ). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for FETZIMA and any potential adverse effects on the breastfed child from FETZIMA or from the underlying maternal conditions.
Clinical Considerations
Infants exposed to FETZIMA should be monitored for agitation, irritability, poor feeding and poor weight gain.
Data
Milnacipran, a racemic mixture that contains levomilnacipran (the 1S,2R-enantiomer of milnacipran), is present in the milk of lactating women treated with milnacipran. In a lactation pharmacokinetic study with milnacipran, a single, oral dose of 50 mg milnacipran HCl tablet was administered to 8 lactating women who were at least 12 weeks postpartum and weaning their infants. The milk/plasma AUC ratio of milnacipran was 1.85 ± 0.38. The maximum estimated weight-adjusted daily infant dose for milnacipran from breast milk (assuming mean milk consumption of 150 mL/kg/day) was 5% of the maternal weight-adjusted dose based on peak plasma concentrations.
8.000000000000000e+00 4 Pediatric Use
The safety and effectiveness of FETZIMA have not been established in pediatric patients for the treatment of major depressive disorder (MDD).
The safety and efficacy of FETZIMA were evaluated in two randomized, double-blind, placebo- and active-controlled 8-week trials in pediatric patients with MDD, one in patients 7 to 17 years of age (flexible-dose study) and the other in patients 12 to 17 years of age (fixed-dose study). The primary efficacy endpoint for both studies was the change from baseline to week 8 in the Children’s Depression Rating Scale-Revised (CDRS-R) total score. The CDRS-R assesses the severity of depression and change in depressive symptoms in children and adolescents with depression. FETZIMA was not superior to placebo in either study. The most commonly observed adverse reactions in pediatric patients 7 to 17 years of age randomized to FETZIMA were similar to those observed in adults [see Adverse Reactions (6.1 )].
FETZIMA was associated with an increase in blood pressure in placebo- and active-controlled trials in pediatric patients with MDD. Increases in blood pressure in pediatric patients treated with FETZIMA led to a higher proportion of pediatric patients developing new-onset and sustained hypertension when compared to adults [see Warnings and Precautions (5.3 )].
Antidepressants increase the risk of suicidal thoughts and behaviors in pediatric patients [ see Boxed Warning, Warnings and Precautions (5.1 ), and Adverse Reactions (6.1 ) ] .
Juvenile Animal Toxicity Data
In a juvenile animal study, male and female rats were treated with 10, 35, or 120 mg/kg/day of levomilnacipran by oral gavage from post-natal day 21 to 90. At 120 mg/kg/day, there was a decrease in bone mineral density in both males and females and a decrease in mean tibia length in females. These effects were not completely resolved at the end of the recovery period. There was a delay in sexual maturation in females treated with 120 mg/kg/day; however, there was no effect on fertility. The no observed adverse effect level (NOAEL) for all these findings was 35 mg/kg/day.
8.000000000000000e+00 5 Geriatric Use
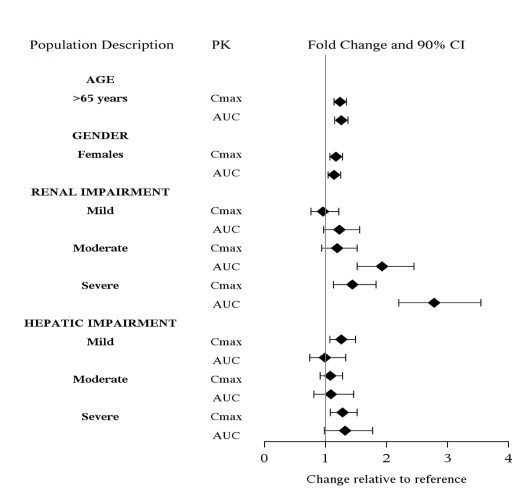
No dose adjustment is recommended on the basis of age [ see Clinical Pharmacology (12.3 ) ] .
Of the total number of patients in the 8-week clinical studies of FETZIMA, 2.8% of patients were age 65 or older.
Because levomilnacipran is predominantly excreted by the kidney, renal clearance of levomilnacipran should be considered when determining the dose [ see Dosage and Administration (2.3 ) ] .
SNRIs, including FETZIMA, have been associated with cases of clinically significant hyponatremia in elderly patients, who may be at greater risk for this adverse reaction [see Warnings and Precautions (5.11 )].
8.000000000000000e+00 6 Hepatic Impairment
Dose adjustment is not recommended in patients with mild (Child-Pugh score of 1-6), moderate (Child-Pugh score of 7-9), or severe (Child-Pugh score of 10-13) hepatic impairment [see Clinical Pharmacology (12.3 )].
8.000000000000000e+00 7 Renal Impairment
Renal excretion plays a predominant role in the elimination of levomilnacipran.
FETZIMA is not recommended for use in patients with end stage renal disease.
Dosing adjustment is recommended for patients with moderate (creatinine clearance of 30 to 59 mL/min) or severe (creatinine clearance of 15 to 29 mL/min) renal impairment [see Dosage and Administration (2.3 ) and Clinical Pharmacology (12.3 ) ] .
Dose adjustment is not recommended for patients with mild (creatinine clearance of 60 to 89 mL/min) renal impairment [see Clinical Pharmacology (12.3 )] .
4 CONTRAINDICATIONS
FETZIMA is contraindicated:
- in patients with hypersensitivity to levomilnacipran, milnacipran HCl, or to any excipient in the formulation.
- with the use of MAOIs intended to treat psychiatric disorders with FETZIMA or within 7 days of stopping treatment with FETZIMA is contraindicated because of an increased risk of serotonin syndrome. The use of FETZIMA within 14 days of stopping an MAOI intended to treat psychiatric disorders is also contraindicated [see Dosage and Administration (2.5 ) and Warnings and Precautions (5.2 )] .
Starting FETZIMA in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome [see Dosage and Administration (2.6 ) and Warnings and Precautions (5.2 )] .
5 WARNINGS AND PRECAUTIONS
- Serotonin Syndrome: Increased risk when co-administered with other serotonergic agents, but also when taken alone. If it occurs, discontinue FETZIMA and serotonergic agents and initiate supportive treatment (5.2 ).
- Elevated Blood Pressure and Heart Rate : Control hypertension before initiating therapy with FETZIMA. Monitor blood pressure regularly during treatment (5.3 , 5.4 ).
- Increased Risk of Bleeding : Concomitant use of NSAIDs, aspirin, other antiplatelet drugs, warfarin, and other anticoagulants may increase this risk (5.5 ).
- A ngle C losure Glaucoma : Angle closure glaucoma has occurred in patients with untreated anatomically narrow angles treated with antidepressants (5.6 ).
- Urinary Hesitation or Retention : Can occur. If such symptoms occur, discontinue FETZIMA or consider other appropriate medical intervention (5.7 ).
- Activation of Mania/Hypomania : Screen patients for bipolar disorder. Caution patients about risk of activation of mania/hypomania (5.8 ).
- Seizures: Can occur. Use with caution in patients with a seizure disorder (5.9 ).
- Discontinuation Syndrome : Taper dose when possible and monitor for discontinuation symptoms (5.10 ).
- Hyponatremia : Can occur in association with SIADH (5.11 ).
- Sexual Dysfunction: FETZIMA may cause symptoms of sexual dysfunction (5.12 ) .
5.1 Suicidal Thoug hts and Behaviors in Adolescents and Young Adults
In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and 4,500 pediatric patients, the incidence of suicidal thoughts and behaviors in antidepressant-treated patients aged 24 years and younger was greater than in placebo-treated patients. There was considerable variation in risk of suicidal thoughts and behaviors among drugs, but there was an increased risk identified in young patients for most drugs studied. There were differences in absolute risk of suicidal thoughts and behaviors across the different indications, with the highest incidence in patients with MDD. The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1000 patients treated are provided in Table 1.
| Age Range | Drug-Placebo Difference in Number of Patients of Suicidal Thoughts or Behaviors per 1000 Patients Treated |
| Increases Compared to Placebo | |
| <18 years old | 14 additional patients |
| 18-24 years old | 5 additional patients |
| Decreases Compared to Placebo | |
| 25-64 years old | 1 fewer patient |
| ≥65 years old | 6 fewer patients |
•Fetzima is not approved for use in pediatric patients.
It is unknown whether the risk of suicidal thoughts and behaviors in children, adolescents, and young adults extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with MDD that antidepressants delay the recurrence of depression and that depression itself is a risk factor for suicidal thoughts and behaviors.
Monitor all antidepressant-treated patients for any indication for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of drug therapy, and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behavior and to alert the healthcare provider. Consider changing the therapeutic regimen, including possibly discontinuing FETZIMA, in patients whose depression is persistently worse, or who are experiencing emergent suicidal thoughts or behaviors.
5.2 Serotonin Syndrome
Serotonin-norepinephrine reuptake inhibitors (SNRIs), including FETZIMA, can precipitate serotonin syndrome, a potentially life-threatening condition. The risk is increased with concomitant use of other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, meperidine, methadone, tryptophan, buspirone, amphetamines, and St. John’s Wort) and with drugs that impair metabolism of serotonin, i.e., MAOIs [see Contraindications (4 ), Drug Interactions (7.1 )]. Serotonin syndrome can also occur when these drugs are used alone.
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
The concomitant use of FETZIMA with MAOIs is contraindicated. In addition, do not initiate FETZIMA in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection). If it is necessary to initiate treatment with a MAOI such as linezolid or intravenous methylene blue in a patient taking FETZIMA, discontinue FETZIMA before initiating treatment with the MAOI [see Dosage and Administration (2.5 , 2.6 ) and Contraindications (4 ), Drug Interactions (7.1 )].
Monitor all patients taking FETZIMA for the emergence of serotonin syndrome. Discontinue treatment with FETZIMA and any concomitant serotonergic agents immediately if the above events occur and initiate supportive symptomatic treatment. If concomitant use of FETZIMA with other serotonergic drugs is clinically warranted, inform patients of the increased risk for serotonin syndrome and monitor for symptoms.
5.3 Elevated Blood Pressure
SNRIs, including FETZIMA, have been associated with increases in blood pressure. Blood pressure should be measured prior to initiating treatment and periodically throughout FETZIMA treatment. Pre-existing hypertension should be controlled before initiating treatment with FETZIMA. Caution should be exercised in treating patients with pre-existing hypertension, cardiovascular, or cerebrovascular conditions that might be compromised by increases in blood pressure. For patients who experience a sustained increase in blood pressure while receiving FETZIMA, discontinuation or other appropriate medical intervention should be considered.
Table 2 shows the mean changes in blood pressure, sustained hypertension, and upward shifts in hypertensive status that were observed in FETZIMA-treated adult patients in the short-term placebo-controlled studies.
| Placebo | FETZIMA40to120 mg/day | ||
| Mean change from baseline to end of treatment, mm Hg | |||
| Systolic blood pressure (SBP) | -0.4 | 3.0 | |
| Diastolic blood pressure (DBP) | 0.0 | 3.2 | |
| Sustained Hypertension, % of patients | |||
| Broad Criteria: SBP ≥ 140 mm Hg and an increase ≥15 mm Hg OR DBP ≥ 90 mm Hg and an increase ≥ 10 mm Hg for at least 3 consecutive visits | 1.2 | 1.8 | |
| Strict Criteria: SBP ≥ 140 mm Hg and an increase ≥15 mm Hg AND DBP ≥ 90 mm Hg and an increase ≥ 10 mm Hg for at least 3 consecutive visits | 0.1 | 0.3 | |
| Upward Shifts in Hypertensive Status a , % of patients | |||
| Normal/ Pre-hypertensive → Stage I/ Stage II | 7.1 | 10.4 | |
| a Normal Blood Pressure: SBP < 120 mm Hg andDBP < 80 mm Hg Pre-hypertension: SBP ≥ 120 mm Hg and≤ 139 mm Hg or DBP ≥ 80 mm Hg and≤ 89 mm Hg Stage I hypertension: SBP ≥ 140 mm Hg and≤ 159 mm Hg or DBP ≥ 90 mm Hg and≤ 99 mm Hg Stage II hypertension: SBP ≥ 160 mm Hg orDBP ≥ 100 mm Hg | |||
In the short-term, placebo-controlled MDD studies in adults, the mean increase from initiation of treatment in systolic BP was 3 mm Hg and diastolic BP was 3.2 mm Hg, as compared to no change in the placebo group. There were no dose-related changes in systolic and diastolic blood pressure observed.
In adult patients exposed to one-year, open-label treatment of FETZIMA (doses range from 40-120 mg once daily), the mean change from initiation of treatment in systolic BP was 3.9 mm Hg and diastolic BP was 3.1 mm Hg.
In short-term, placebo- and active-controlled MDD studies in pediatric patients 7 years to less than 18 years of age, treatment with FETZIMA was associated with the occurrence of new-onset hypertension (two systolic and/or diastolic BP measurements in the stage I hypertension range and/or one measurement in the stage II range) in 36.2% of treated patients compared with 20.7% of patients randomized to placebo. Elevations in either systolic or diastolic BP leading to measures at or above the stage II hypertension threshold occurred in 12.1% of pediatric patients treated with FETZIMA and 7.5% of patients randomized to placebo. Sustained hypertension (three or more consecutive systolic or diastolic BP measurements at or above the stage I hypertension threshold) occurred in 15% of pediatric patients treated with FETZIMA and 4% of patients randomized to placebo. The safety and effectiveness of FETZIMA have not been established in pediatric patients for the treatment of MDD.
In the short-term, placebo-controlled studies in adults, 11.6% of patients met orthostatic hypotension criteria (SBP or DBP) in the FETZIMA group compared to 9.7% in the placebo group. Orthostatic reductions of blood pressure ≥ 10 mm Hg in DBP occurred in 5.8%, 6.1%, and 9.8% of FETZIMA-treated patients with doses of 40, 80, and 120 mg/day respectively, compared to 6.2% of placebo-treated patients.
Concomitant use of FETZIMA with drugs that increase blood pressure and heart rate has not been evaluated and such combinations should be used with caution. Effects of FETZIMA on blood pressure in patients with significant hypertension or cardiac disease have not been systematically evaluated. FETZIMA should be used with caution in these patients.
5.4 Elevated Heart Rate
SNRIs, including FETZIMA, have been associated with increased heart rate. Heart rate should be measured prior to initiating treatment and periodically throughout FETZIMA treatment. Pre-existing tachyarrhythmias and other cardiac disease should be treated before starting therapy with FETZIMA. For patients who experience a sustained increase in heart rate while receiving FETZIMA, discontinuation or other appropriate medical intervention should be considered.
In short-term clinical studies, FETZIMA treatment was associated with a mean increase in heart rate of 7.4 beats per minute (bpm) compared to a mean decrease of 0.3 bpm in placebo-treated patients. Heart rate increase in FETZIMA-treated patients receiving doses of 40 mg, 80 mg, and 120 mg was 7.2, 7.2, and 9.1 bpm.
FETZIMA has not been systematically evaluated in patients with a cardiac rhythm disorder.
5.5 Increased Risk of Bleeding
Drugs that interfere with serotonin reuptake, including FETZIMA, may increase the risk of bleeding events. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), warfarin, and other anticoagulants may add to this risk. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. Based on data from the published observational studies, exposure to SNRIs, particularly in the month before delivery, has been associated with a less than 2-fold increase in the risk of postpartum hemorrhage [see Use in Specific Populations (8.1 )]. Bleeding events related to SSRIs and SNRIs have ranged from ecchymosis, hematoma, epistaxis, and petechiae to life-threatening hemorrhages.
Inform patients about the increased risk of bleeding associated with the concomitant use of FETZIMA and NSAIDs, aspirin, or other drugs that affect coagulation [see Drug Interactions (7.1 )] .
5.6 Angle Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs including FETZIMA may trigger an angle closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy. Avoid use of antidepressants, including FETZIMA, in patients with anatomically narrow angles.
5.7 Urinary Hesitation or Retention
The noradrenergic effect of SNRIs including FETZIMA, can affect urethral resistance. In the controlled short-term studies, urinary hesitation occurred in 4%, 5%, and 6% of FETZIMA-treated patients receiving doses of 40, 80, and 120 mg, respectively, compared to no patients in the placebo group. Caution is advised in the use of FETZIMA in patients prone to obstructive urinary disorders. If symptoms of urinary hesitation, urinary retention, or dysuria develop during treatment with FETZIMA, consideration should be given to the possibility that they might be drug-related, and discontinuation or other appropriate medical intervention should be considered.
5.000000000000000e+00 8 Activation of Mania/Hypomania
Symptoms of mania/hypomania were reported in 0.2% of FETZIMA-treated patients and 0.2% of placebo-treated patients in clinical studies. Activation of mania/hypomania has also been reported in a small proportion of patients with mood disorders who were treated with other antidepressants. Prior to initiating treatment with FETZIMA, screen patients for any personal or family history of bipolar disorder, mania, or hypomania.
5.000000000000000e+00 9 Seizures
One case of seizure has been reported in pre-marketing clinical studies with FETZIMA. FETZIMA has not been systematically evaluated in patients with a seizure disorder. Patients with a history of seizures were excluded from clinical studies. FETZIMA should be prescribed with caution in patients with a seizure disorder.
5.000000000000000e+00 10 Discontinuation Syndrome
There have been reports of adverse events occurring upon discontinuation of serotonergic antidepressants, particularly when discontinuation is abrupt, including the following: dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paresthesia, such as electric shock sensations), anxiety, confusion, headache, lethargy, emotional lability, insomnia, hypomania, tinnitus, and seizures. While these events are generally self-limiting, there have been reports of serious discontinuation symptoms.
Monitor patients for these symptoms when discontinuing FETZIMA. Reduce the dose gradually whenever possible. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, consider resuming the previously prescribed dose. Subsequently, the dose may be decreased, but at a more gradual rate [see Dosage and Administration (2.4 )] .
5.1 1 Hyponatremia
Hyponatremia may occur as a result of treatment with SSRIs and SNRIs, including FETZIMA. In many cases, hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Cases with serum sodium lower than 110 mmol/L have been reported. Elderly patients may be at greater risk of developing hyponatremia with SSRIs and SNRIs. Also, patients taking diuretics or who are otherwise volume depleted can be at greater risk. FETZIMA should be discontinued in patients with symptomatic hyponatremia and appropriate medical intervention should be instituted. Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which can lead to falls. Signs and symptoms associated with more severe and/or acute cases have included hallucination, syncope, seizure, coma, respiratory arrest, and death.
5.1 2 Sexual Dysfunction
Use of SNRIs, including FETZIMA, may cause symptoms of sexual dysfunction [see Adverse Reactions (6.1 )] . In male patients, SNRI use may result in ejaculatory delay or failure, decreased libido, and erectile dysfunction. In female patients, SNRI use may result in decreased libido and delayed or absent orgasm.
It is important for prescribers to inquire about sexual function prior to initiation of FETZIMA and to inquire specifically about changes in sexual function during treatment, because sexual function may not be spontaneously reported. When evaluating changes in sexual function, obtaining a detailed history (including timing of symptom onset) is important because sexual symptoms may have other causes, including the underlying psychiatric disorder. Discuss potential management strategies to support patients in making informed decisions about treatment.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label.
- Hypersensitivity [see Contraindications (4 )]
- Suicidal Thoughts and Behaviors in Adolescents and Young Adults [see Warnings and Precautions (5.1 )]
- Serotonin Syndrome [see Warnings and Precautions (5.2 )]
- Elevated Blood Pressure [see Warnings and Precautions (5.3 )]
- Elevated Heart Rate [see Warnings and Precautions (5.4 )]
- Increased Risk of Bleeding [see Warnings and Precautions (5.5 )]
- Angle Closure Glaucoma [see Warnings and Precautions (5.6 )]
- Urinary Hesitation or Retention [see Warnings and Precautions (5.7 )]
- Activation of Mania/Hypomania [see Warnings and Precautions (5.8 )]
- Seizure [see Warnings and Precautions (5.9 )]
- Discontinuation Syndrome [see Warnings and Precautions (5.10 )]
- Hyponatremia [see Warnings and Precautions (5.11 )]
- Sexual Dysfunction [see Warnings and Precautions (5.12 )]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Patient e xposure
The safety of FETZIMA was evaluated in 3,317 patients (18 to 78 years of age) diagnosed with MDD who participated in clinical studies, representing 1,186 patient-years of exposure. Among the 3,317 FETZIMA-treated patients, 1,583 were exposed to FETZIMA in short-term, placebo-controlled studies. There were 825 patients who continued from short-term studies into a one-year, open-label extension study.
Of the 3,317 patients exposed to at least one dose of FETZIMA, 895 patients were exposed to FETZIMA for at least 6 months and 367 were exposed for one year. In these studies, FETZIMA was given at doses ranging from 40 mg to 120 mg once daily and was given without regard to food.
Adverse r eacti ons r eported as r easons for discontinuation of t reatment
In the short-term placebo-controlled pre-marketing studies for MDD, 9% of the 1,583 patients who received FETZIMA (40 mg to 120 mg) discontinued treatment due to an adverse reaction, compared with 3% of the 1,040 placebo-treated patients in those studies. The most common adverse reaction leading to discontinuation in at least 1% of the FETZIMA-treated patients in the short-term placebo-controlled studies was nausea (1.5%).
Common a dverse r eactions in p lacebo- c ontrolled MDD s tudies
The most commonly observed adverse reactions in FETZIMA-treated MDD patients in placebo-controlled studies (incidence ≥ 5% and at least twice the rate of placebo) were: nausea, constipation, hyperhidrosis, heart rate increased, erectile dysfunction, ejaculation disorder, tachycardia, vomiting, and palpitations.
Table 3 shows the incidence of adverse reactions that occurred in ≥ 2% of FETZIMA-treated MDD patients and at least twice the rate of placebo in the placebo-controlled studies.
| System Organ Class Preferred Term | Placebo(N =1040)% | FETZIMA 40mg to120 mgper day(N = 1583) % |
| Gastrointestinal disorders | ||
| Nausea | 6 | 17 |
| Constipation | 3 | 9 |
| Vomiting | 1 | 5 |
| Cardiac disorders | ||
| Tachycardia a | 2 | 6 |
| Palpitations | 1 | 5 |
| Reproductive system and breast disorders b | ||
| Erectile dysfunction c | 1 | 6 |
| Testicular pain d | <1 | 4 |
| Ejaculation disorder e | <1 | 5 |
| Investigations | ||
| Heart rate increased f | 1 | 6 |
| Blood pressure increased g | 1 | 3 |
| Renal and urinary disorders | ||
| Urinary hesitation | 0 | 4 |
| Skin and s ubcutaneous t issue d isorders | ||
| Hyperhidrosis | 2 | 9 |
| Rash h | 0 | 2 |
| Vascular disorders | ||
| Hot flush | 1 | 3 |
| Hypotension i | 1 | 3 |
| Hypertension j | 1 | 3 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 1 | 3 |
| a Tachycardia also includes: sinus tachycardia and postural orthostatic tachycardia syndrome b Percentage is relative to the number of patients in the associated demographic sex category. Fewer than 2% of FETZIMA-treated MDD female patients in placebo-controlled clinical studies reported adverse events related to sexual function. c Erectile dysfunction includes: erectile dysfunction, organic erectile dysfunction, and psychogenic erectile dysfunction d Testicular pain includes: testicular pain, epididymitis, and seminal vesiculitis e Ejaculation disorder includes: ejaculation disorder, ejaculation delayed, ejaculation failure, and premature ejaculation f Heart rate increased also includes: orthostatic heart rate response increased g Blood pressure increased also includes: blood pressure systolic increased, blood pressure diastolic increased, and blood pressure orthostatic increased h Rash also includes: rash generalized, rash maculo-papular, rash erythematous, and rash macular i Hypotension also includes: orthostatic hypotension and dizziness postural j Hypertension also includes: labile hypertension N = number of patients in the Safety Population | ||
Dose- r elated a dverse r eactions
In pooled data from the short-term placebo-controlled fixed-dose studies, there were no dose-related adverse reactions (greater than 2% overall incidence) in patients treated with FETZIMA across the dose range 40 mg to 120 mg once daily, with the exception of erectile dysfunction and urinary hesitation (see Table 4).
| System Organ Class Preferred Term | Placebo (N = 362) % | FETZIMA | ||
| 40 mgper day (N = 366) % | 80 mgper day (N = 367) % | 120 mgper day (N = 180) % | ||
| Urinary hesitation | 0 | 4 | 5 | 6 |
| Erectile dysfunction a | 2 | 6 | 8 | 10 |
| a Percentage is relative to the number of male patients. N = number of patients in the Safety Population | ||||
Other adverse reactions observed in clinical studies
Other infrequent adverse reactions, not described elsewhere in the label, occurring at an incidence of < 2% in MDD patients treated with FETZIMA were:
Cardiac disorders: Angina pectoris; Supraventricular and Ventricular extrasystoles
Eye disorders: Dry eye; Vision blurred; Conjunctival hemorrhage
General disorders: Chest pain; Thirst
Gastrointestinal disorders: Abdominal pain; Flatulence
Investigations disorders: Blood cholesterol increased; Liver function test abnormal
Nervous System disorders: Migraine; Paraesthesia; Syncope; Extrapyramidal disorder
Psychiatric disorders: Agitation; Anger; Bruxism; Panic attack; Tension; Aggression
Renal and Urinary disorder: Pollakiuria; Hematuria; Proteinuria
Respiratory, thoracic and mediastinal disorders: Yawning
Skin and subcutaneous tissue disorders: Dry skin; Pruritus; Urticaria
6.2 Post m arketing Experience
The following adverse reaction has been identified during post-approval use of FETZIMA or other selective serotonin and norepinephrine reuptake inhibitors. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac disorders : Takotsubo cardiomyopathy
Respiratory, thoracic and mediastinal disorders : Anosmia, Hyposmia
7 DRUG INTERACTIONS
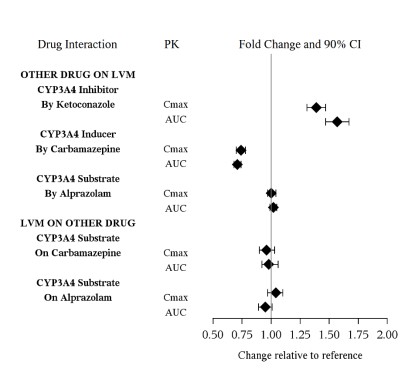
- Strong CYP3A4 inhibitors : Maximum recommended dosage is 80 mg once daily (7 ).
7.1 Drugs Having Clinically Important Interactions with FETZIMA
Table 5 includes clinically important drug interactions with FETZIMA.
| Monoamine Oxidase Inhibitors (MAOIs) | |
| Clinical Impact: | Concomitant use of SSRIs and SNRIs including FETZIMA with MAOIs increases the risk of serotonin syndrome. |
| Intervention: | Concomitant use of FETZIMA is contraindicated:
|
| Examples: | selegiline, tranylcypromine, isocarboxazid, phenelzine, linezolid, methylene blue |
| Other Serotonergic Drugs | |
| Clinical Impact: | Concomitant use of FETZIMA with other serotonergic drugs increases the risk of serotonin syndrome. |
| Intervention: | Monitor for symptoms of serotonin syndrome when FETZIMA is used concomitantly with other drugs that may affect the serotonergic neurotransmitter systems. If serotonin syndrome occurs, immediately discontinue FETZIMA and/or concomitant serotonergic drugs [ see Dosage and Administration (2.5 , 2.6 ) , Contraindications (4 ) , and Warnings and Precautions (5.2 ) ] . |
| Examples: | other SNRIs, SSRIs, triptans, tricyclic antidepressants, opioids, lithium, buspirone, amphetamines, tryptophan, and St. John's Wort |
| Drugs that Interfere with Hemostasis | |
| Clinical Impact: | Concomitant use of FETZIMA with an antiplatelet or anticoagulant drug may potentiate the risk of bleeding. This may be due to the effect of FETZIMA on the release of serotonin by platelets. |
| Intervention: | Closely monitor for bleeding for patients receiving an antiplatelet or anticoagulant drug when FETZIMA is initiated or discontinued [see Warnings and Precautions (5.5 )] . |
| Examples: | NSAIDs, aspirin, and warfarin |
| Strong CYP3A4 Inhibitors | |
| Clinical Impact: | Concomitant use of FETZIMA with strong CYP3A4 inhibitors increases levomilnacipran exposure [see Pharmacokinetics (12.3 )] . |
| Intervention: | The dose of FETZIMA should not exceed 80 mg once daily when used with strong CYP3A4 inhibitors [see Dosage and Administration (2.6 ). |
| Examples: | Ketoconazole, itraconazole, clarithromycin |
| Alcohol | |
| Clinical Impact: | Concomitant use of FETZIMA and alcohol may result in accelerated release of levomilnacipran. |
| Intervention: | Avoid concomitant use of FETZIMA and alcohol [see Clinical Pharmacology (12.3 )] . |
11 DESCRIPTION
FETZIMA contains levomilnacipran, a selective serotonin and norepinephrine reuptake inhibitor (SNRI), in the form of hydrochloride salt with the chemical name of levomilnacipran hydrochloride is (1S,2R)-2-(aminomethyl)-N,N-diethyl-1-phenylcyclopropanecarboxamide hydrochloride. Its empirical formula is C 15 H 2 2 N 2 O∙HCl and its molecular weight is 282.8 g/mol. Levomilnacipran (Initial US approval: 2013) is the 1S,2R-enantiomer of milnacipran. The chemical structure of levomilnacipran hydrochloride is:
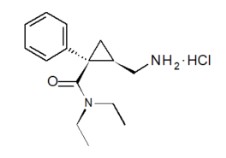
FETZIMA extended-release capsules are intended for oral administration. Each FETZIMA capsule contains 23.0, 45.9, 91.8, or 137.8 mg of levomilnacipran hydrochloride equivalent to 20, 40, 80, or 120 mg of levomilnacipran, respectively.
Inactive ingredients include ethylcellulose, hypromellose, povidone, sugar spheres, talc, titanium dioxide, and triethyl citrate. Inactive ingredients also include black iron oxide, red iron oxide (80 mg and 120 mg capsules only), shellac glaze, and yellow iron oxide (20 mg and 40 mg capsules only).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The exact mechanism of the antidepressant action of levomilnacipran is unknown, but is thought to be related to the potentiation of serotonin and norepinephrine in the central nervous system, through inhibition of reuptake at serotonin and norepinephrine transporters. Non-clinical studies have shown that levomilnacipran is a potent and selective serotonin and norepinephrine reuptake inhibitor (SNRI).
12.2 Pharmacodynamics
Levomilnacipran binds with high affinity to the human serotonin (5-HT) and norepinephrine (NE) transporters (Ki = 11 and 91 nM, respectively) and potently inhibits 5-HT and NE reuptake (IC50 = 16-19 and 11 nM, respectively). Levomilnacipran lacks significant affinity for any other receptors, ion channels or transporters tested in vitro , including serotonergic (5HT1-7), α- and β- adrenergic, muscarinic, or histaminergic receptors and Ca 2+ , Na+, K+ or Cl- channels. Levomilnacipran did not inhibit monoamine oxidase (MAO).
Cardiovascular Electrophysiology
At a dose 2.5 times the maximum recommended dose, levomilnacipran does not prolong QTc to any clinically relevant extent.
12.3 Pharmacokinetics
The concentration of levomilnacipran at steady state is proportional to dose when administered from 25 mg to 300 mg (2.5 times the maximum recommended dosage of FETZIMA) once daily. Steady-state concentrations of levomilnacipran are predictable from single-dose data. After daily dosing of FETZIMA 120 mg, the mean C max value is 341 ng/mL, and the mean steady-state AUC value is 5196 ng·h/mL. Interconversion between levomilnacipran and its stereoisomer does not occur in humans.
Absorption
The relative bioavailability of levomilnacipran after administration of FETZIMA was 92% when compared to oral solution. The median time to peak concentration (T max ) of levomilnacipran is 6-8 hours after oral administration.
Effect of Food
Levomilnacipran concentration was not significantly affected when FETZIMA was administered with food.
Distribution
Levomilnacipran is widely distributed with an apparent volume of distribution of 387-473 L; plasma protein binding is 22% over concentration range of 10 to 1000 ng/mL.
Elimination
Following an oral administration, the mean apparent total clearance of levomilnacipran is 21-29 L/h. The apparent terminal elimination half-life of levomilnacipran is approximately 12 hours.
Metabolism
Levomilnacipran undergoes desethylation to form desethyl levomilnacipran and hydroxylation to form p-hydroxy-levomilnacipran. Both oxidative metabolites undergo further conjugation with glucuronide to form conjugates. The desethylation is catalyzed primarily by CYP3A4 with minor contribution by CYP2C8, 2C19, 2D6, and 2J2.
Excretion
Levomilnacipran and its metabolites are eliminated primarily by renal excretion. Following oral administration of 14C-levomilnacipran solution, approximately 58% of the dose is excreted in urine as unchanged levomilnacipran. N-desethyl levomilnacipran is the major metabolite excreted in the urine and accounted for approximately 18% of the dose. Other identifiable metabolites excreted in the urine are levomilnacipran glucuronide (4%), desethyl levomilnacipran glucuronide (3%), p-hydroxy levomilnacipran glucuronide (1%), and p-hydroxy levomilnacipran (1%). The metabolites are inactive [see Dosage and Administration (2.3 )] .
Drug Interaction Studies
Clinical Studies
The drug interaction studies for levomilnacipran are summarized in Figure 1.
Figure 1. PK Interactions between Levomilnacipran (LVM) and Other Drugs
In vitro Studies
In vitro studies suggested that CYP2C8, CYP2C19, CYP2D6, and CYP2J2 had minimal contributions to metabolism of levomilnacipran. In addition, levomilnacipran is not a substrate of BCRP, OATP1B1, OATP1B3, OAT1, OAT3, or OCT2 and is a weak substrate of P-gp.
In vitro studies have shown that levomilnacipran is not an inhibitor of CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, P-gp, OATP1B1, OATP1B3, OAT1, OAT3, or OCT2.
Alcohol
An in vitro study indicated increases of levomilnacipran release from FETZIMA extended-release capsules (20, 40, 80, and 120 mg) at 2 hours by approximately 9.5%, 23%, and 56% in the presence of 5%, 20%, and 40% (v/v) alcohol, respectively. Effect of 40% alcohol resulted in nearly complete drug release in 4 hours. There is no in vivo study conducted for the effect of alcohol on drug exposure.
Speci fic Populations
The effect of intrinsic patient factors on the pharmacokinetics of levomilnacipran is presented in Figure 2.
Figure 2 Effect of Intrinsic Factors on Levomilnacipran PK
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Levomilnacipran administered by oral gavage to rats for 2 years and Tg.rasH2 mice for 6 months did not increase the incidence of tumors in either study.
Rats received levomilnacipran at doses up to 90/70 mg/kg/day (the dose was lowered in males after 45 weeks of dosing). The 90 mg/kg/day dose is 7 times the maximum recommended human dose (MRHD) of 120 mg on a mg/m 2 basis.
Tg.rasH2 mice received levomilnacipran at doses up to 150 mg/kg/day.
Mutagenesis
Levomilnacipran was not mutagenic in the in vitro bacterial mutation assay (Ames test) and was not clastogenic in an in vivo micronucleus assay in rats. Additionally, levomilnacipran was not genotoxic in the in vitro mouse lymphoma (L5178Y TK+/-) cell forward mutation assay.
Impairment of Fertility
When levomilnacipran was administered orally to male and female rats before mating, through mating and up to Day 7 of gestation at doses up to 100 mg/kg/day, no effects were observed on fertility. This dose is 8 times the MRHD on a mg/m 2 basis.
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Patient e xposure
The safety of FETZIMA was evaluated in 3,317 patients (18 to 78 years of age) diagnosed with MDD who participated in clinical studies, representing 1,186 patient-years of exposure. Among the 3,317 FETZIMA-treated patients, 1,583 were exposed to FETZIMA in short-term, placebo-controlled studies. There were 825 patients who continued from short-term studies into a one-year, open-label extension study.
Of the 3,317 patients exposed to at least one dose of FETZIMA, 895 patients were exposed to FETZIMA for at least 6 months and 367 were exposed for one year. In these studies, FETZIMA was given at doses ranging from 40 mg to 120 mg once daily and was given without regard to food.
Adverse r eacti ons r eported as r easons for discontinuation of t reatment
In the short-term placebo-controlled pre-marketing studies for MDD, 9% of the 1,583 patients who received FETZIMA (40 mg to 120 mg) discontinued treatment due to an adverse reaction, compared with 3% of the 1,040 placebo-treated patients in those studies. The most common adverse reaction leading to discontinuation in at least 1% of the FETZIMA-treated patients in the short-term placebo-controlled studies was nausea (1.5%).
Common a dverse r eactions in p lacebo- c ontrolled MDD s tudies
The most commonly observed adverse reactions in FETZIMA-treated MDD patients in placebo-controlled studies (incidence ≥ 5% and at least twice the rate of placebo) were: nausea, constipation, hyperhidrosis, heart rate increased, erectile dysfunction, ejaculation disorder, tachycardia, vomiting, and palpitations.
Table 3 shows the incidence of adverse reactions that occurred in ≥ 2% of FETZIMA-treated MDD patients and at least twice the rate of placebo in the placebo-controlled studies.
| System Organ Class Preferred Term | Placebo(N =1040)% | FETZIMA 40mg to120 mgper day(N = 1583) % |
| Gastrointestinal disorders | ||
| Nausea | 6 | 17 |
| Constipation | 3 | 9 |
| Vomiting | 1 | 5 |
| Cardiac disorders | ||
| Tachycardia a | 2 | 6 |
| Palpitations | 1 | 5 |
| Reproductive system and breast disorders b | ||
| Erectile dysfunction c | 1 | 6 |
| Testicular pain d | <1 | 4 |
| Ejaculation disorder e | <1 | 5 |
| Investigations | ||
| Heart rate increased f | 1 | 6 |
| Blood pressure increased g | 1 | 3 |
| Renal and urinary disorders | ||
| Urinary hesitation | 0 | 4 |
| Skin and s ubcutaneous t issue d isorders | ||
| Hyperhidrosis | 2 | 9 |
| Rash h | 0 | 2 |
| Vascular disorders | ||
| Hot flush | 1 | 3 |
| Hypotension i | 1 | 3 |
| Hypertension j | 1 | 3 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 1 | 3 |
| a Tachycardia also includes: sinus tachycardia and postural orthostatic tachycardia syndrome b Percentage is relative to the number of patients in the associated demographic sex category. Fewer than 2% of FETZIMA-treated MDD female patients in placebo-controlled clinical studies reported adverse events related to sexual function. c Erectile dysfunction includes: erectile dysfunction, organic erectile dysfunction, and psychogenic erectile dysfunction d Testicular pain includes: testicular pain, epididymitis, and seminal vesiculitis e Ejaculation disorder includes: ejaculation disorder, ejaculation delayed, ejaculation failure, and premature ejaculation f Heart rate increased also includes: orthostatic heart rate response increased g Blood pressure increased also includes: blood pressure systolic increased, blood pressure diastolic increased, and blood pressure orthostatic increased h Rash also includes: rash generalized, rash maculo-papular, rash erythematous, and rash macular i Hypotension also includes: orthostatic hypotension and dizziness postural j Hypertension also includes: labile hypertension N = number of patients in the Safety Population | ||
Dose- r elated a dverse r eactions
In pooled data from the short-term placebo-controlled fixed-dose studies, there were no dose-related adverse reactions (greater than 2% overall incidence) in patients treated with FETZIMA across the dose range 40 mg to 120 mg once daily, with the exception of erectile dysfunction and urinary hesitation (see Table 4).
| System Organ Class Preferred Term | Placebo (N = 362) % | FETZIMA | ||
| 40 mgper day (N = 366) % | 80 mgper day (N = 367) % | 120 mgper day (N = 180) % | ||
| Urinary hesitation | 0 | 4 | 5 | 6 |
| Erectile dysfunction a | 2 | 6 | 8 | 10 |
| a Percentage is relative to the number of male patients. N = number of patients in the Safety Population | ||||
Other adverse reactions observed in clinical studies
Other infrequent adverse reactions, not described elsewhere in the label, occurring at an incidence of < 2% in MDD patients treated with FETZIMA were:
Cardiac disorders: Angina pectoris; Supraventricular and Ventricular extrasystoles
Eye disorders: Dry eye; Vision blurred; Conjunctival hemorrhage
General disorders: Chest pain; Thirst
Gastrointestinal disorders: Abdominal pain; Flatulence
Investigations disorders: Blood cholesterol increased; Liver function test abnormal
Nervous System disorders: Migraine; Paraesthesia; Syncope; Extrapyramidal disorder
Psychiatric disorders: Agitation; Anger; Bruxism; Panic attack; Tension; Aggression
Renal and Urinary disorder: Pollakiuria; Hematuria; Proteinuria
Respiratory, thoracic and mediastinal disorders: Yawning
Skin and subcutaneous tissue disorders: Dry skin; Pruritus; Urticaria
16 HOW SUPPLIED/STORAGE AND HANDLING
FETZIMA extended-release capsules are supplied in the following configurations:
| Capsule Strength | Capsule Color/Shape | Capsule Markings | Package Configuration | NDC Code |
| 20 mg | yellow cap white body | black "FL" on cap black "20" on body | Bottle / 30 count | 0456-2220-30 |
| Hospital Unit Dose (Blister) / 10 x 10 | 0456-2220-63 | |||
| 40 mg | yellow cap yellow body | black "FL" on cap black "40" on body | Bottle / 30 count | 0456-2240-30 |
| Bottle / 90 count | 0456-2240-90 | |||
| Hospital Unit Dose (Blister) / 10 x 10 | 0456-2240-63 | |||
| 80 mg | pink cap white body | black "FL" on cap black "80" on body | Bottle / 30 count | 0456-2280-30 |
| Bottle / 90 count | 0456-2280-90 | |||
| Hospital Unit Dose (Blister) /10 x 10 | 0456-2280-63 | |||
| 120 mg | pink cap pink body | black "FL" on cap black "120" on body | Bottle / 30 count | 0456-2212-30 |
| Bottle / 90 count | 0456-2212-90 | |||
| Hospital Unit Dose (Blister) / 10 x 10 | 0456-2212-63 |
FETZIMA Titration Pack is supplied in the following configuration:
| Capsule Strength | Capsule Color/Shape | Capsule Markings | Package Configuration | NDC Code |
| 20 mg | yellow cap white body | black "FL" on cap black "20" on body | Titration Pack (Blister) containing two 20 mg capsules and twenty-six 40 mg capsules | 0456-2202-28 |
| 40 mg | yellow cap yellow body | black "FL" on cap black "40" on body |
Storage and Handling
All package configurations: Store at 25°C (77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [See USP Controlled Room Temperature] .
12.1 Mechanism of Action
The exact mechanism of the antidepressant action of levomilnacipran is unknown, but is thought to be related to the potentiation of serotonin and norepinephrine in the central nervous system, through inhibition of reuptake at serotonin and norepinephrine transporters. Non-clinical studies have shown that levomilnacipran is a potent and selective serotonin and norepinephrine reuptake inhibitor (SNRI).