Get your patient on Gemtesa (Vibegron)
Gemtesa patient education
Patient toolkit
Dosage & administration

Gemtesa prescribing information
INDICATIONS AND USAGE
GEMTESA is a beta-3 adrenergic agonist indicated for the treatment of:
- overactive bladder (OAB) with symptoms of urge urinary incontinence, urgency, and urinary frequency in adults. (1.1 )
- overactive bladder (OAB) with symptoms of urge urinary incontinence, urgency, and urinary frequency in adult males on pharmacological therapy for benign prostatic hyperplasia (BPH). (1.2 )
Overactive Bladder in Adults
GEMTESA ® is indicated for the treatment of overactive bladder (OAB) with symptoms of urge urinary incontinence, urgency, and urinary frequency in adults.
Overactive Bladder in Adult Males with Benign Prostatic Hyperplasia (BPH)
GEMTESA is indicated for the treatment of overactive bladder (OAB) with symptoms of urge urinary incontinence, urgency, and urinary frequency in adult males on pharmacological therapy for benign prostatic hyperplasia (BPH).
DOSAGE AND ADMINISTRATION
Recommended Dosage
The recommended dosage of GEMTESA is one 75 mg tablet orally, once daily with or without food. Swallow GEMTESA tablets whole with a glass of water.
In adults, GEMTESA tablets also may be crushed, mixed with a tablespoon (approximately 15 mL) of applesauce and taken immediately with a glass of water [see Clinical Pharmacology (12.3 )] .
DOSAGE FORMS AND STRENGTHS
Tablets: 75 mg, oval, light green, film-coated, debossed with V75 on one side and no debossing on the other side.
USE IN SPECIFIC POPULATIONS
Pediatric use : Safety and effectiveness in pediatric patients have not been established. (8.4 )
End-stage Renal Disease with or without Hemodialysis : Not recommended. (8.6 )
Severe Hepatic Impairment : Not recommended. (8.7 )
Pregnancy
Risk Summary
There are no available data on GEMTESA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
In animal studies, no effects on embryo-fetal development were observed following administration of vibegron during the period of organogenesis at exposures approximately 275-fold and 285-fold greater than clinical exposure at the recommended daily dose of GEMTESA, in rats and rabbits, respectively. Delayed fetal skeletal ossification was observed in rabbits at approximately 898-fold clinical exposure, in the presence of maternal toxicity. In rats treated with vibegron during pregnancy and lactation, no effects on offspring were observed at 89-fold clinical exposure. Developmental toxicity was observed in offspring at approximately 458-fold clinical exposure, in the presence of maternal toxicity. No effects on offspring were observed at 89-fold clinical exposure (see Data ).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies carry some risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal developmental toxicity study, pregnant rats were treated with daily oral doses of 0, 30, 100, 300, or 1000 mg/kg/day vibegron during the period of organogenesis (Days 6 to 20 of gestation). These doses were associated with systemic exposures (AUC) 0-, 9-, 89-, 275-, and 1867-fold higher, respectively, than in humans treated with the recommended daily dose of GEMTESA. No embryo-fetal developmental toxicity was observed at doses up to 300 mg/kg/day. Treatment with the high dose of 1000 mg/kg/day was discontinued due to maternal toxicity.
In an embryo-fetal developmental toxicity study, pregnant rabbits were treated with daily oral doses of 0, 30, 100, or 300 mg/kg/day vibegron during the period of organogenesis (Days 7 to 20 of gestation). These doses were associated with systemic exposures (AUC) 0-, 86-, 285-, and 898-fold higher, respectively, than in humans treated with the recommended daily dose of GEMTESA. No embryo-fetal developmental toxicity was observed at doses of vibegron up to 100 mg/kg/day. Maternal toxicity (decreased food consumption), reduced fetal body weight, and an increased incidence of delayed skeletal ossification, were observed at 300 mg/kg/day.
In a pre- and post-natal developmental toxicity study, pregnant or lactating rats were treated with daily oral doses of 0, 30, 100, or 500 mg/kg/day vibegron from day 6 of gestation through day 20 of lactation. These doses were associated with estimated systemic exposures (AUC) 0-, 9-, 89-, and 458-fold higher, respectively, than in humans treated with the recommended daily dose of GEMTESA. No developmental toxicity was observed in F1 offspring at doses up to 100 mg/kg/day. Maternal toxicity was observed during lactation (decreased body weight gain) at doses ≥100 mg/kg/day and during gestation (decreased body weight gain and food consumption) at 500 mg/kg/day. Developmental toxicity was observed in F1 offspring (increased stillborn index, lethality, reduced viability and weaning indices, decreased body weight and body weight gains, low physical development differentiation indices, and effects on sensory function and reflexes) at 500 mg/kg/day.
Lactation
Risk Summary
There are no data on the presence of vibegron in human milk, the effects of the drug on the breastfed infant, or the effects on milk production. When a single oral dose of radiolabeled vibegron was administered to postnatal nursing rats, radioactivity was observed in milk (see Data ). When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for GEMTESA and any potential adverse effects on the breastfed infant from GEMTESA or from the underlying maternal condition.
Data
Animal Data
In a lactational transfer study, lactating rats were treated with a single oral dose of 10 mg/kg radiolabeled [ 3 H] vibegron on postpartum day 10. Levels of radioactivity were determined in milk and plasma collected at 1, 4, 12, and 24 after dosing. The C max of total radioactivity in milk and plasma were observed at 9 and 2 hours after dosing, respectively, with a maximum milk-to-plasma concentration ratio of 2.2 observed at 12 hours after dosing. Vibegron elimination from milk showed a similar trend as that from plasma. The radioactivity concentration in milk at 24 hours after administration was approximately 25% of the C max .
Pediatric Use
The safety and effectiveness of GEMTESA in pediatric patients have not been established.
Geriatric Use
Of 526 patients who received GEMTESA in the clinical studies for OAB with symptoms of urge urinary incontinence, urgency, and urinary frequency, 242 (46%) were 65 years of age or older, and 75 (14%) were 75 years of age or older [see Clinical Studies (14.1 )] . No overall differences in safety or effectiveness of GEMTESA have been observed between patients 65 years of age and older and younger adult patients.
Of the total number of GEMTESA-treated patients in clinical studies for OAB with symptoms of urge urinary incontinence, urgency, and urinary frequency in adult males on pharmacological therapy for benign prostatic hyperplasia (BPH), 347 (63%) were 65 years of age and older, while 100 (18%) were 75 years of age and older [see Clinical Studies (14.2 )] . No overall differences in safety of GEMTESA have been observed between patients 65 years of age and older and younger adult patients.
Renal Impairment
No dosage adjustment for GEMTESA is recommended for patients with mild, moderate, or severe renal impairment (eGFR 15 to <90 mL/min/1.73 m 2 ). GEMTESA has not been studied in patients with eGFR <15 mL/min/1.73 m 2 (with or without hemodialysis) and is not recommended in these patients [see Clinical Pharmacology (12.3 )] .
Hepatic Impairment
No dosage adjustment for GEMTESA is recommended for patients with mild to moderate hepatic impairment (Child-Pugh A and B). GEMTESA has not been studied in patients with severe hepatic impairment (Child-Pugh C) and is not recommended in this patient population [see Clinical Pharmacology (12.3 )] .
WARNINGS AND PRECAUTIONS
Urinary Retention : Monitor for urinary retention, especially in patients with bladder outlet obstruction and also in patients taking muscarinic antagonist medications for OAB, in whom the risk of urinary retention may be greater. If urinary retention develops, discontinue GEMTESA. (5.1 )
Angioedema : Angioedema of the face and/or larynx has been reported with GEMTESA. (5.2 )
Urinary Retention
Urinary retention has been reported in patients taking GEMTESA. The risk of urinary retention may be increased in patients with bladder outlet obstruction and also in patients taking muscarinic antagonist medications for the treatment of OAB. Monitor patients for signs and symptoms of urinary retention, particularly in patients with bladder outlet obstruction or patients taking muscarinic antagonist medications for the treatment of OAB. Discontinue GEMTESA in patients who develop urinary retention [see Adverse Reactions (6.1 )] .
Angioedema
Angioedema of the face and/or larynx has been reported with GEMTESA. Angioedema has been reported to occur hours after the first dose or after multiple doses. Angioedema, associated with upper airway swelling, may be life-threatening. If involvement of the tongue, hypopharynx, or larynx occurs, immediately discontinue GEMTESA and provide appropriate therapy and/or measures necessary to ensure a patent airway. GEMTESA is contraindicated in patients with known hypersensitivity to vibegron or any component of GEMTESA [see Contraindications (4 ) and Adverse Reactions (6.2 )] .
ADVERSE REACTIONS
The following clinically significant adverse reaction is described elsewhere in the labeling:
- Urinary retention [see Warnings and Precautions (5.1 )]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Overactive Bladder in Adults
The safety of GEMTESA was evaluated in a 12-week, double-blind, placebo- and active-controlled study (Study 3003) in patients with OAB [see Clinical Studies (14.1 )] . A total of 545 patients received GEMTESA. The majority of the patients were White (78%) and female (85%) with a mean age of 60 years (range 18 to 93 years).
Adverse reactions that were reported in Study 3003 at an incidence greater than placebo and in ≥2% of patients treated with GEMTESA are listed in Table 1 .
| GEMTESA 75 mg n (%) | Placebo n (%) | |
|---|---|---|
| Number of Patients | 545 | 540 |
| Headache | 22 (4.0) | 13 (2.4) |
| Nasopharyngitis | 15 (2.8) | 9 (1.7) |
| Diarrhea | 12 (2.2) | 6 (1.1) |
| Nausea | 12 (2.2) | 6 (1.1) |
| Upper respiratory tract infection | 11 (2.0) | 4 (0.7) |
Other adverse reactions reported in <2% of patients treated with GEMTESA included:
Gastrointestinal disorders: dry mouth, constipation
Investigations: residual urine volume increased
Renal and urinary disorders: urinary retention
Vascular disorders: hot flush
GEMTESA was also evaluated for long-term safety in an extension study (Study 3004) in 505 patients who completed the 12-week study (Study 3003). Of the 273 patients who received GEMTESA 75 mg once daily in the extension study, 181 patients were treated for a total of one year.
Adverse reactions reported in ≥2% of patients treated with GEMTESA 75 mg for up to 52 weeks in the long-term extension study, and not already listed above, were urinary tract infection (6.6%) and bronchitis (2.9%).
Overactive Bladder in Adult Males with BPH
The safety of GEMTESA was evaluated in a 24-week double-blind, randomized, placebo-controlled study (Study 3005) in male patients with OAB on pharmacological therapy for BPH. A total of 553 patients received GEMTESA [see Clinical Studies (14.2 )] .
Adverse reactions that were reported in Study 3005 at an incidence greater than placebo and in ≥2% of patients treated with GEMTESA are listed in Table 2 .
| GEMTESA 75 mg n (%) | Placebo n (%) | |
|---|---|---|
•Defined as an average systolic blood pressure (SBP) ≥140mmHg or diastolic BP (DBP) ≥90mmHg on 3 assessments at two consecutive visits, in non-hypertensive patients. | ||
•Defined as an average increase of SBP ≥20mmHg or DBP≥10mmHg on 3 assessments at two consecutive visits, or the initiation or increase in dose of antihypertensive medications at any visit, in hypertensive patients. | ||
| Number of Patients | 553 | 551 |
| Hypertension• | 50 (9.0) | 46 (8.3) |
| Urinary tract infection | 14 (2.5) | 12 (2.2) |
GEMTESA was also evaluated for long-term safety in a 28-week extension study (Study 3006) in 276 patients who completed the 24-week study (Study 3005). Of the 276 patients who received GEMTESA 75 mg once daily in the extension study, 124 patients were treated for a total of one year.
There were no additional adverse reactions reported in Study 3006 that are not already included in Section 6.1 above.
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of vibegron. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following adverse events have been reported in association with vibegron use in worldwide postmarketing experience:
Urologic disorders : urinary retention
Skin and subcutaneous tissue disorders : angioedema of the face and larynx; hypersensitivity reactions, including urticaria, pruritus, rash and drug eruption; eczema
Gastrointestinal disorders : constipation
DRUG INTERACTIONS
Concomitant use of GEMTESA increases digoxin maximal concentrations (C max ) and systemic exposure as assessed by area under the concentration-time curve (AUC) [see Clinical Pharmacology (12.3 )] . Serum digoxin concentrations should be monitored before initiating and during therapy with GEMTESA and used for titration of the digoxin dose to obtain the desired clinical effect. Continue monitoring digoxin concentrations upon discontinuation of GEMTESA and adjust digoxin dose as needed.
DESCRIPTION
Vibegron is a selective beta-3 adrenergic agonist. The chemical name is (6S)-N-[4-[[(2S,5R)-5-[(R)-hydroxy(phenyl)methyl]pyrrolidin-2-yl]methyl]phenyl]-4-oxo-7,8-dihydro-6H-pyrrolo[1,2-a]pyrimidine-6-carboxamide having a molecular formula of C 26 H 28 N 4 O 3 and a molecular weight of 444.538 g/mol. The structural formula of vibegron is:
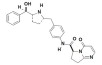
Vibegron is a crystalline, white to off-white to tan powder.
GEMTESA tablets, for oral administration contain 75 mg of vibegron and the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium stearate, mannitol, and microcrystalline cellulose. The light green film coating contains FD&C Blue No. 2 - aluminum lake, hypromellose, iron oxide yellow, lactose monohydrate, titanium dioxide, and triacetin.
CLINICAL PHARMACOLOGY
Mechanism of Action
Vibegron is a selective human beta-3 adrenergic receptor agonist. Activation of the beta-3 adrenergic receptor increases bladder capacity by relaxing the detrusor smooth muscle during bladder filling.
Pharmacodynamics
Vibegron's exposure-response relationship and the time course of pharmacodynamic response are not fully characterized.
Blood Pressure
In a 4-week, randomized, placebo-controlled, ambulatory blood pressure study in OAB patients (n=200), daily treatment with GEMTESA 75 mg was not associated with clinically significant changes in blood pressure. Subjects enrolled in this study had a mean age of 59 years and 75% were female. Thirty-five percent of subjects had pre-existing hypertension at baseline and 29% of all subjects were taking at least 1 concomitant antihypertensive medication.
Cardiac Electrophysiology
GEMTESA does not prolong the QT interval to any clinically relevant extent at a single dose 5.3 times the approved recommended dose.
Pharmacokinetics
Mean vibegron C max and AUC increased in a greater than dose-proportional manner up to 600 mg (8 times the approved recommended dosage). Steady state concentrations are achieved within 7 days of once daily dosing. The mean accumulation ratio (R ac ) was 1.7 for C max and 2.4 for AUC 0-24hr .
Absorption
Median vibegron T max is approximately 1 to 3 hours.
Oral administration of a 75 mg vibegron tablet crushed and mixed with 15 mL of applesauce resulted in no clinically relevant changes in vibegron pharmacokinetics when compared to administration of an intact 75 mg vibegron tablet.
Effect of Food
No clinically significant differences in vibegron pharmacokinetics were observed following administration of a high-fat meal (53% fat, 869 calories [32.1 g protein, 70.2 g carbohydrate, and 51.1 g fat]).
Distribution
The mean apparent volume of distribution is 6304 liters. Human plasma protein binding of vibegron is approximately 50%. The average blood-to-plasma concentration ratio is 0.9.
Elimination
Vibegron has an effective half-life of 30.8 hours across all populations.
Metabolism
Metabolism plays a minor role in the elimination of vibegron. CYP3A4 is the predominant enzyme responsible for in vitro metabolism.
Excretion
Following a radiolabeled dose, approximately 59% of the dose (54% as unchanged) was recovered in feces and 20% (19% as unchanged) in urine.
Specific Populations
No clinically significant differences in the pharmacokinetics of vibegron were observed based on age (18 to 93 years), sex, race/ethnicity (Japanese vs. non-Japanese), mild (eGFR 60 to <90 mL/min/1.73 m 2 ), moderate (eGFR 30 to <60 mL/min/1.73 m 2 ), and severe (eGFR 15 to <30 mL/min/1.73 m 2 ) renal impairment, or moderate (Child-Pugh B) hepatic impairment. The effect of more severe renal impairment (eGFR <15 mL/min/1.73 m 2 ) with or without hemodialysis or severe (Child-Pugh C) hepatic impairment on vibegron pharmacokinetics was not studied.
Drug Interaction Studies
Clinical Studies
Digoxin : Concomitant administration of vibegron increased digoxin C max and AUC by 21% and 11%, respectively.
Other Drugs: No clinically significant differences in vibegron pharmacokinetics were observed when used concomitantly with ketoconazole (P-gp and strong CYP3A4 inhibitor), diltiazem (P-gp and moderate CYP3A4 inhibitor), rifampin (strong CYP3A4 inducer), or tolterodine. No clinically significant differences in the pharmacokinetics of the following drugs were observed when used concomitantly with vibegron: tolterodine, tolterodine 5-hydroxy metabolite, metoprolol, combined oral contraceptive (ethinyl estradiol, levonorgestrel), or warfarin.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Vibegron is a CYP3A4 substrate. Vibegron did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4. Vibegron did not induce CYP1A2, CYP2B6, or CYP3A4.
Transporter Systems: Vibegron is a P-gp substrate. Vibegron did not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2K at clinically relevant concentrations.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No carcinogenicity was observed in long-term studies conducted in mice and rats treated with daily oral doses of vibegron for approximately 2 years. In the mouse carcinogenicity study, CD-1 mice were treated with daily oral doses of vibegron up to 90 mg/kg/day in males and up to 150 mg/kg/day in females, corresponding to estimated systemic exposures (AUC) 21- and 55-fold higher, respectively, than in humans treated with the recommended daily dose of GEMTESA. In the rat carcinogenicity study, Sprague Dawley rats were treated with daily oral doses of vibegron up to 30 mg/kg/day in males and up to 180 mg/kg/day in females, corresponding to systemic exposures (AUC) 18- and 117-fold higher, respectively, than in humans treated with the recommended daily dose of GEMTESA.
Mutagenesis
Vibegron was not mutagenic in in vitro microbial reverse mutation assays, showed no evidence of genotoxic activity in an in vitro human peripheral blood lymphocyte chromosomal aberration assay, and did not increase the frequency of micronucleated polychromatic erythrocytes in an in vivo rat bone marrow micronucleus assay.
Impairment of Fertility
In fertility/general reproductive toxicity studies conducted in rats, females were treated with daily oral doses of 0, 30, 100, 300, or 1000 mg/kg/day vibegron and males were treated with daily oral doses of 0, 10, 30, or 300 mg/kg/day vibegron. No effects on fertility were observed in female or male rats at doses up to 300 mg/kg/day, associated with systemic exposure (AUC) at least 274-fold higher than in humans treated with the recommended daily dose of GEMTESA. General toxicity, decreased fecundity, and decreased fertility were observed in female rats at 1000 mg/kg/day, associated with estimated systemic exposure 1867-fold higher than in humans treated with the recommended daily dose of GEMTESA.
CLINICAL STUDIES
Overactive Bladder in Adults
The efficacy of GEMTESA was evaluated in a 12-week, double-blind, randomized, placebo-controlled, and active-controlled trial (Study 3003, NCT03492281) in patients with OAB (urge urinary incontinence, urgency, and urinary frequency). Patients were randomized 5:5:4 to receive either GEMTESA 75 mg, placebo, or active control orally, once daily for 12 weeks. For study entry, patients had to have symptoms of OAB for at least 3 months with an average of 8 or more micturitions per day and at least 1 urge urinary incontinence (UUI) per day, or an average of 8 or more micturitions per day and an average of at least 3 urgency episodes per day. Urge urinary incontinence was defined as leakage of urine of any amount because the patient felt an urge or need to urinate immediately. The study population included OAB medication-naïve patients as well as patients who had received prior therapy with OAB medications.
The co-primary endpoints were change from baseline in average daily number of micturitions and average daily number of UUI episodes at week 12. Additional endpoints included change from baseline in average daily number of “need to urinate immediately” (urgency) episodes and average volume voided per micturition.
A total of 1,515 patients received at least one daily dose of placebo (n=540), GEMTESA 75 mg (n=545), or an active control treatment (n=430). The majority of patients were White (78%) and female (85%) with a mean age of 60 (range 18 to 93) years.
Table 3 shows changes from baseline at week 12 for average daily number of micturitions, average daily number of UUI episodes, average daily number of “need to urinate immediately” (urgency) episodes, and average volume voided per micturition.
| Parameter | GEMTESA 75 mg | Placebo |
|---|---|---|
• Least squares mean adjusted for treatment, baseline, sex, geographical region, study visit, and study visit by treatment interaction term. | ||
| Average Daily Number of Micturitions | ||
| Baseline mean (n) | 11.3 (526) | 11.8 (520) |
| Change from Baseline • (n) | -1.8 (492) | -1.3 (475) |
| Difference from Placebo | -0.5 | |
| 95% Confidence Interval | -0.8, -0.2 | |
| p-value | <0.001 | |
| Average Daily Number of UUI Episodes | ||
| Baseline mean (n) | 3.4 (403) | 3.5 (405) |
| Change from Baseline • (n) | -2.0 (383) | -1.4 (372) |
| Difference from Placebo | -0.6 | |
| 95% Confidence Interval | -0.9, -0.3 | |
| p-value | <0.0001 | |
| Average Daily Number of “Need to Urinate Immediately” (Urgency) Episodes | ||
| Baseline mean (n) | 8.1 (526) | 8.1 (520) |
| Change from Baseline • (n) | -2.7 (492) | -2.0 (475) |
| Difference from Placebo | -0.7 | |
| 95% Confidence Interval | -1.1, -0.2 | |
| p-value | 0.002 | |
| Average Volume Voided (mL) per Micturition | ||
| Baseline mean (n) | 155 (524) | 148 (514) |
| Change from Baseline • (n) | 23 (490) | 2 (478) |
| Difference from Placebo | 21 | |
| 95% Confidence Interval | 14, 28 | |
| p-value | <0.0001 | |
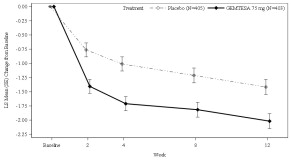
Figure 1 and Figure 2 show the mean change from baseline over time in average daily number of micturitions and mean change from baseline over time in average daily number of UUI episodes, respectively.
Figure 1: Mean (SE) Change from Baseline in the Average Daily Number of Micturitions
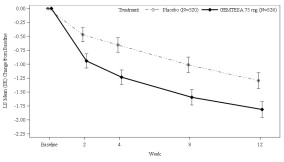
Figure 2: Mean (SE) Change from Baseline in the Average Daily Number of UUI Episodes in Patients with At Least 1 Average Daily UUI Episode at Baseline
Overactive Bladder in Adult Males with BPH
The safety, tolerability, and efficacy of GEMTESA was evaluated in a multinational 24-week, double-blind, randomized, placebo-controlled trial (Study 3005, NCT03902080) in male patients at least 45 years of age with OAB on pharmacological therapy (i.e., treatment with an alpha blocker, with or without a 5-alpha reductase inhibitor) for BPH. A total of 1105 patients were randomized 1:1 to receive either GEMTESA 75 mg or placebo once daily for 24 weeks. For study entry, patients had symptoms of OAB (an average of 8 or more micturitions per day, 3 or more urgency episodes per day with or without incontinence, and 2 or more nocturia episodes per night) while taking pharmacological therapy for at least 2 months for the treatment of lower urinary tract symptoms due to BPH. Randomization was stratified based on the baseline average number of micturition episodes per day, alpha blocker use with or without 5 alpha reductase inhibitor use, and urinary incontinence.
The co-primary endpoints were change from baseline in the average daily number of micturitions and the average daily number of “need to urinate immediately” (urgency) episodes at week 12. Additional endpoints included change from baseline in the average daily number of urge urinary incontinence (UUI) episodes and the average volume voided per micturition.
A total of 1104 patients received at least one daily dose of placebo (n=551) or GEMTESA 75 mg (n=553). The majority of patients were White (87%) and enrolled in the U.S. (56%). The mean age was 67 years (range 45 to 97) and at least 63% were ≥ 65 years.
Table 4 shows changes from baseline at week 12 for average daily number of micturitions, average daily number of urgency episodes, average daily number of UUI episodes and average volume voided per micturition.
| Parameter | GEMTESA 75 mg (N = 538) | Placebo (N = 542) |
|---|---|---|
• Least squares mean adjusted for study visit, baseline value, geographical region, interaction of visit by treatment, and stratification factors as randomized (baseline average micturitions per day••, alpha blocker use with or without 5-ARI, baseline urinary incontinence••), geographical region, study visit, and study visit by treatment interaction term. | ||
•• Baseline average micturitions per day stratification factor is not included in the model where the continuous value of baseline average micturitions per day is present; baseline urinary incontinence is not included in the model for UUI; | ||
••• Only subjects with baseline incontinence have UUI analyzed. | ||
| Average Daily Number of Micturitions | ||
| Baseline mean | 11.84 | 11.96 |
| Change from Baseline • | -2.04 | -1.30 |
| Difference from Placebo | -0.74 | |
| 95% Confidence Interval | -1.02, -0.46 | |
| Average Daily Number of “Need to Urinate Immediately” (Urgency) Episodes | ||
| Baseline mean | 9.05 | 9.00 |
| Change from Baseline • | -2.88 | -1.93 |
| Difference from Placebo | -0.95 | |
| 95% Confidence Interval | -1.37, -0.54 | |
| Average Daily Number of UUI Episodes | ||
| N ••• | 146 | 151 |
| Baseline mean | 3.33 | 3.23 |
| Change from Baseline • | -2.19 | -1.39 |
| Difference from Placebo | -0.80 | |
| 95% Confidence Interval | -1.33, -0.27 | |
| Average Volume Voided (mL) per Micturition | ||
| Baseline mean | 166.37 | 166.43 |
| Change from Baseline • | 25.63 | 10.56 |
| Difference from Placebo | 15.07 | |
| 95% Confidence Interval | 9.13, 21.02 | |
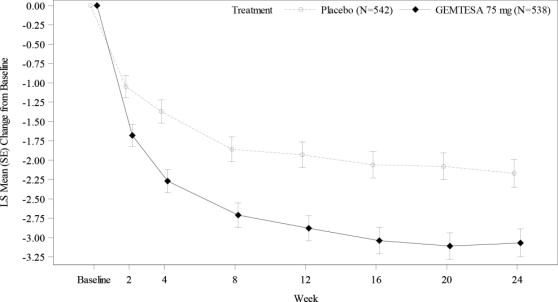
Figure 3 and Figure 4 show the mean change from baseline over time in average daily number of micturitions and mean change from baseline over time in average daily number of urgency episodes, respectively.
Figure 3: Mean (SE) Change from Baseline in the Average Daily Number of Micturitions in Male Patients with BPH on Pharmacological Therapy
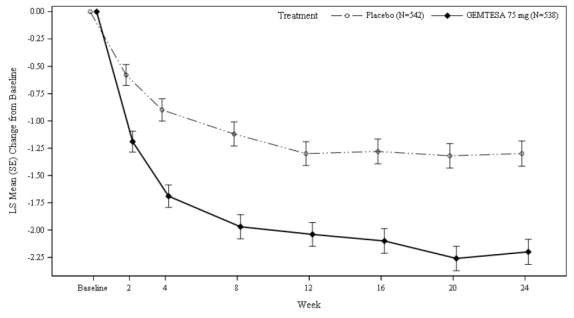
Figure 4: Mean (SE) Change from Baseline in the Average Daily Number of Urgency Episodes in Male Patients with BPH on Pharmacological Therapy
HOW SUPPLIED/STORAGE AND HANDLING
GEMTESA 75 mg tablets are light green, oval, film-coated tablets, debossed with V75 on one side and no debossing on the other side.
GEMTESA is marketed in two packaging configurations:
Thirty (30) tablets in a HDPE bottle with a child-resistant cap, NDC 73336-075-30
Ninety (90) tablets in a HDPE bottle with a child-resistant cap, NDC 73336-075-90
Store at 20°C to 25°C (68°F to 77°F), excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Keep this and all medications out of sight and reach of children.
Dispose unused medication via a take-back option if available; otherwise follow FDA instructions for disposal in the household trash. See www.fda.gov/drugdisposal for more information.
Mechanism of Action
Vibegron is a selective human beta-3 adrenergic receptor agonist. Activation of the beta-3 adrenergic receptor increases bladder capacity by relaxing the detrusor smooth muscle during bladder filling.