Get your patient on Inrebic (Fedratinib Hydrochloride)
Inrebic patient education
Patient toolkit
Dosage & administration

Inrebic prescribing information
WARNING: ENCEPHALOPATHY INCLUDING WERNICKE'S
Serious and fatal encephalopathy, including Wernicke's, has occurred in patients treated with INREBIC. Wernicke's encephalopathy is a neurologic emergency. Assess thiamine levels in all patients prior to starting INREBIC. Do not start INREBIC in patients with thiamine deficiency; replete thiamine prior to treatment initiation. While on treatment all patients should receive prophylaxis with daily oral thiamine and should have thiamine levels assessed as clinically indicated. If encephalopathy is suspected, immediately discontinue INREBIC and initiate parenteral thiamine. Monitor until symptoms resolve or improve and thiamine levels normalize [see Dosage and Administration (2.6) , Warnings and Precautions (5.1) and Adverse Reactions (6.1) ] .
INDICATIONS AND USAGE
INREBIC ® is indicated for the treatment of adult patients with intermediate-2 or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis (MF).
DOSAGE AND ADMINISTRATION
- Recommended Dosage: 400 mg orally once daily with or without food for patients with a baseline platelet count of greater than or equal to 50 × 10 9 /L (2.2 ).
- For patients who have difficulty swallowing capsules whole or those with a nasogastric tube, the content of the capsule(s) may be dispersed in Ensure ® Plus (2.2 ).
- Reduce dose for patients taking strong CYP3A inhibitors or with severe renal impairment (2.3 , 2.4 , 7.1 , 8.6 ).
Required Concomitant Medications
During treatment with INREBIC, all patients should receive prophylaxis with thiamine 100 mg orally daily [see Dosage and Administration (2.7) and Warnings and Precautions (5.1) ].
Recommended Dosage
Conduct baseline testing of thiamine (Vitamin B1) levels prior to initiation of INREBIC [see Dosage and Administration (2.7) , Warnings and Precautions (5.1) ] .
The recommended dosage of INREBIC is 400 mg taken orally once daily for patients with a baseline platelet count of greater than or equal to 50 × 10 9 /L.
Modify the dose for patients using concomitant strong CYP3A4 inhibitors, and in patients with severe renal impairment (creatinine clearance (CL cr ) 15 mL/min to 29 mL/min) [see Dosage and Administration (2.4 , 2.5) ] .
Patients that are on treatment with ruxolitinib before the initiation of INREBIC must taper and discontinue according to the ruxolitinib prescribing information.
Administration Information:
- INREBIC may be taken with or without food. Administration with a high fat meal may reduce the incidence of nausea and vomiting.
- If a dose of INREBIC is missed, the next scheduled dose should be taken the following day.
- For patients who have difficulty swallowing capsule(s) whole or those with a nasogastric tube:
- Open the capsule(s).
- In a glass container, mix the content of the capsule(s) with approximately 180 mL of Ensure ® Plus liquid nutritional supplement [see Clinical Pharmacology (12.3) ] at room temperature [between 20°C to 25°C (68°F to 77°F)].
- Promptly administer the mixture orally or through a nasogastric tube (French size 14 or 16) within 2 hours of preparation.
- If using a nasogastric tube, flush it with 60 mL of water after administering the mixture [see Clinical Pharmacology (12.3 )].
- Discard the prepared dose if not given within 2 hours.
Monitoring for Safety
Obtain the following blood tests prior to starting treatment with INREBIC, periodically during treatment, and as clinically indicated [see Warnings and Precautions (5.1 , 5.2 , 5.4 , 5.5) ]:
- Thiamine (Vitamin B1) level
- Complete blood count with platelets
- Creatinine and BUN
- Hepatic panel
- Amylase and lipase
Dose Modifications with Concomitant Use of Strong CYP3A4 Inhibitors
Reduce INREBIC dose when administering with strong CYP3A4 inhibitors to 200 mg once daily.
In cases where coadministration with a strong CYP3A4 inhibitor is discontinued, INREBIC dosage should be increased to 300 mg once daily during the first two weeks after discontinuation of the CYP3A4 inhibitor, and then to 400 mg once daily thereafter as tolerated [see Drug Interactions (7.1) ] .
Dose Modifications for Severe Renal Impairment
Reduce INREBIC dose to 200 mg once daily in patients with severe renal impairment (creatinine clearance (CL cr ) 15 mL/min to 29 mL/min as estimated by Cockcroft-Gault (C-G) equation).
Dose Modifications for Adverse Reactions
Modify dose for hematologic and nonhematologic adverse reactions per Table 1 and Table 2. Discontinue INREBIC in patients unable to tolerate a dose of 200 mg daily. See Warnings and Precautions for other mitigating strategies.
Hematologic Adverse Reactions | Dose Reduction |
Grade 4 Thrombocytopenia or Grade 3 Thrombocytopenia with active bleeding | Interrupt dose until resolved to Grade 2 or lower or baseline. Restart dose at 100 mg daily below the last given dose. |
Grade 4 Neutropenia | Interrupt dose until resolved to Grade 2 or lower or baseline. Restart dose at 100 mg daily below the last given dose. |
Consider dose reductions for patients who become transfusion-dependent during treatment with INREBIC.
Nonhematologic Adverse Reactions | Dose Reduction |
Grade 3 or higher Nausea, Vomiting, or Diarrhea not responding to supportive measures within 48 hours | Interrupt dose until resolved to Grade 1 or lower or baseline. Restart dose at 100 mg daily below the last given dose. |
Grade 3 or higher ALT, AST, or Bilirubin | Interrupt dose until resolved to Grade 1 or lower or baseline. Restart dose at 100 mg daily below the last given dose. Monitor ALT, AST, and bilirubin (total and direct) more frequently following the dose reduction. If reoccurrence of a Grade 3 or higher elevation, discontinue treatment with INREBIC. |
Grade 3 or higher Other Nonhematologic Toxicities | Interrupt dose until resolved to Grade 1 or lower or baseline. Restart dose at 100 mg daily below the last given dose. |
Management of Thiamine Levels and Wernicke's Encephalopathy (WE)
Assess thiamine levels and nutritional status prior to starting INREBIC. Do not start INREBIC in patients with thiamine deficiency. However, if thiamine levels are low, replete thiamine prior to starting treatment. While on INREBIC treatment all patients should receive prophylaxis with daily 100 mg oral thiamine and should have thiamine levels assessed as clinically indicated. If Wernicke's encephalopathy is suspected, immediately discontinue treatment with INREBIC and initiate parenteral thiamine treatment. Monitor until symptoms resolve or improve and thiamine levels normalize [see Warnings and Precautions (5.1) and Adverse Reactions (6.1) ] .
DOSAGE FORMS AND STRENGTHS
Capsules: 100 mg, reddish brown, opaque size 0, printed with "FEDR 100 mg" in white ink.
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed (8.2 ).
Pregnancy
Risk Summary
There are no available data on INREBIC use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, oral administration of fedratinib to pregnant rats during organogenesis at doses considerably lower than the recommended human daily dose of 400 mg/day resulted in adverse developmental outcomes (see Data ) . Consider the benefits and risks of INREBIC for the mother and possible risks to the fetus when prescribing INREBIC to a pregnant woman.
The background risk of major birth defects and miscarriage for the indicated population is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in pregnant rats, fedratinib administration at a dose of 30 mg/kg/day during organogenesis (gestation days 6 to 17) was associated with adverse developmental outcomes including skeletal variations (such as additional ossification center of neuronal arches). These effects occurred in rats at approximately 0.1 times the clinical exposure based on AUC at the recommended daily dose. At lower doses of 10 mg/kg/day (0.01 times the clinical exposure at the recommended daily dose), fedratinib administered to pregnant rats resulted in maternal toxicity of decreased gestational weight gain.
In an embryo-fetal development study in pregnant rabbits, fedratinib administration during organogenesis (gestation Days 6 to 18) did not produce developmental or maternal toxicity at doses up to the highest dose level tested, 30 mg/kg/day (approximately 0.08 times the clinical exposure at the recommended daily dose). In a separate study, administration of 80 mg/kg/day fedratinib to rabbits resulted in maternal mortality.
In a pre- and postnatal study in rats, fedratinib was administered to pregnant female rats at doses of 3, 10, or 30 mg/kg/day from Day 6 of gestation through Day 20 of lactation, with weaning on Day 21. A slight decrease in maternal body weight gain during gestation occurred at 30 mg/kg/day. The offspring from the high dose (30 mg/kg) had decreased body weight preweaning in both sexes and postweaning through the maturation phase in males. These effects occurred at exposures approximately 0.1 times the clinical exposure at the recommended daily dose.
Lactation
Risk Summary
There are no data on the presence of fedratinib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise patients not to breastfeed during treatment with INREBIC, and for at least 1 month after the last dose.
Pediatric Use
The safety and effectiveness of INREBIC in pediatric patients have not been established.
Geriatric Use
Of the total number of patients with myelofibrosis who received an INREBIC dose of 400 mg in the clinical studies, 47.3% were greater than 65 years of age and 12.3% were greater than 75 years of age. No overall differences in safety or effectiveness of INREBIC were observed between these patients and younger patients.
Renal Impairment
Reduce INREBIC dose when administered to patients with severe renal impairment (CL cr 15 mL/min to 29 mL/min by Cockcroft-Gault) [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3) ] . No modification of the starting dose is recommended for patients with mild to moderate renal impairment (CL cr 30 mL/min to 89 mL/min by Cockcroft-Gault). Due to potential increase of exposure, patients with preexisting moderate renal impairment require more intensive safety monitoring, and if necessary, dose modifications based on adverse reactions [see Dosage and Administration (2.6) ].
CONTRAINDICATIONS
None
WARNINGS AND PRECAUTIONS
- Anemia and Thrombocytopenia: Manage by dose reduction, interruption, or transfusion (5.2 ).
- Gastrointestinal Toxicity: Manage by dose reduction or interruption if patient develops severe diarrhea, nausea, or vomiting. Prophylaxis with antiemetics and treatment with antidiarrhea medications are recommended (5.3 ).
- Hepatic Toxicity: Manage by dose reduction or interruption (5.4 ).
- Amylase and Lipase Elevation: Manage by dose reduction or interruption (5.5 ).
- Uveitis: Monitor for symptoms and refer for ophthalmological evaluation (5.6 ).
- Major Adverse Cardiac Events (MACE): Monitor for development of MACE (5.7 ).
- Thrombosis: Evaluate and treat symptoms of thrombosis promptly (5.8 ).
- Secondary Malignancies: Monitor for development of secondary malignancies, particularly in patients who are current or past smokers (5.9 ).
- Symptom Exacerbation Following Interruption or Discontinuation of Treatment: Manage with supportive care and consider resuming treatment with INREBIC (5.10 ).
Encephalopathy, Including Wernicke's
Serious and fatal encephalopathy, including Wernicke's encephalopathy, has occurred in INREBIC-treated patients. Serious cases were reported in 1.3% (8/608) of patients treated with INREBIC in clinical trials and 0.16% (1/608) of cases were fatal.
Wernicke's encephalopathy is a neurologic emergency resulting from thiamine (Vitamin B1) deficiency. Signs and symptoms of Wernicke's encephalopathy may include ataxia, mental status changes, and ophthalmoplegia (e.g., nystagmus, diplopia). Any change in mental status, confusion, or memory impairment should raise concern for potential encephalopathy, including Wernicke's, and prompt a full evaluation including a neurologic examination, assessment of thiamine levels, and imaging. Assess thiamine levels in all patients prior to starting INREBIC. Do not start INREBIC in patients with thiamine deficiency. However, if thiamine levels are low, replete thiamine prior to starting treatment. While on treatment all patients should receive prophylaxis with oral thiamine and should have thiamine levels assessed as clinically indicated. If encephalopathy is suspected, immediately discontinue INREBIC and initiate parenteral thiamine. Monitor until symptoms resolve or improve and thiamine levels normalize [see Dosage and Administration (2.7) and Adverse Reactions (6.1) ] .
Anemia and Thrombocytopenia
Treatment with INREBIC can cause anemia and thrombocytopenia.
Anemia
New or worsening Grade 3 anemia occurred in 34% of INREBIC-treated patients. The median time to onset of the first Grade 3 anemia was approximately 2 months, with 75% of cases occurring within 3 months. Mean hemoglobin levels reached nadir after 12 to 16 weeks with partial recovery and stabilization after 16 weeks. Red blood cell transfusions were received by 51% of INREBIC-treated patients and permanent discontinuation of INREBIC occurred due to anemia in 1% of patients. Consider dose reduction for patients who become red blood cell transfusion dependent [see Dosage and Administration (2.6) ] .
Thrombocytopenia
New or worsening Grade ≥3 thrombocytopenia during the randomized treatment period occurred in 12% of INREBIC-treated patients. The median time to onset of the first Grade 3 thrombocytopenia was approximately 1 month; with 75% of cases occurring within 4 months. Platelet transfusions were received by 3.1% of INREBIC-treated patients. Permanent discontinuation of treatment due to thrombocytopenia and bleeding that required clinical intervention both occurred in 2.1% of INREBIC-treated patients.
Obtain a complete blood count (CBC) at baseline, periodically during treatment, and as clinically indicated. For Grade 3 thrombocytopenia with active bleeding or Grade 4 thrombocytopenia, interrupt INREBIC until resolved to less than or equal to Grade 2 or baseline. Restart dose at 100 mg daily below the last given dose and monitor platelets as clinically indicated [see Dosage and Administration (2.6) ].
Gastrointestinal Toxicity
Gastrointestinal toxicities are the most frequent adverse reactions in INREBIC-treated patients. During the randomized treatment period, diarrhea occurred in 66% of patients, nausea in 62% of patients, and vomiting in 39% of patients. Grade 3 diarrhea and vomiting occurred in 5% and 3.1% of patients, respectively. The median time to onset of any grade nausea, vomiting, and diarrhea was 1 day, with 75% of cases occurring within 2 weeks of treatment.
Consider providing appropriate prophylactic antiemetic therapy (e.g., 5-HT3 receptor antagonists) during INREBIC treatment. Treat diarrhea with antidiarrheal medications promptly at the first onset of symptoms. For Grade 3 or higher nausea, vomiting, or diarrhea not responsive to supportive measures within 48 hours, interrupt INREBIC until resolved to Grade 1 or less or baseline. Restart dose at 100 mg daily below the last given dose [see Dosage and Administration (2.6 ) ] . Monitor thiamine levels and replete as needed.
Hepatic Toxicity
Elevations of ALT and AST (all grades) during the randomized treatment period occurred in 43% and 40%, respectively, with Grade 3 or 4 in 1% and 0%, respectively, of INREBIC-treated patients. The median time to onset of any grade transaminase elevation was approximately 1 month, with 75% of cases occurring within 3 months.
Monitor hepatic function at baseline, periodically during treatment, and as clinically indicated. For Grade 3 or higher ALT and/or AST elevations (greater than 5 × ULN), interrupt INREBIC dose until resolved to Grade 1 or less or to baseline. Restart dose at 100 mg daily below the last given dose. If reoccurrence of a Grade 3 or higher elevation of ALT/AST, discontinue treatment with INREBIC [see Dosage and Administration (2.6) ].
Amylase and Lipase Elevation
Grade 3 or higher amylase and/or lipase elevations developed in 2% and 10%, respectively, of INREBIC-treated patients. The median time to onset of any grade amylase or lipase elevation was 15 days, with 75% of cases occurring within 1 month of starting treatment. One patient developed pancreatitis in the fedratinib clinical development program (n=608) and pancreatitis resolved with treatment discontinuation.
Monitor amylase and lipase at baseline, periodically during treatment, and as clinically indicated. For Grade 3 or higher amylase and/or lipase elevations, interrupt INREBIC until resolved to Grade 1 or less or to baseline. Restart dose at 100 mg daily below the last given dose [see Dosage and Administration (2.6) ].
Uveitis
Uveitis has been observed in post-approval clinical studies with an overall incidence of 4% (11/251). Among patients with INREBIC-associated uveitis, more than half cases observed were in Japanese patients (55%; 6/11). INREBIC-associated uveitis is a late-onset adverse reaction, with the first episode occurring at a median of 14 months after starting treatment, with a range of 8 to 22 months. Recurrent uveitis was reported in some patients who continued INREBIC. The uveitis episodes varied in severity, with grade 1/2 in 60% of episodes, and grade 3/4 in 40% of episodes. Topical steroids were sufficient for treatment in 75% of episodes, and systemic steroids were required in 25% of episodes. Among the patients developing uveitis, INREBIC was discontinued due to uveitis in 27% of patients.
Advise patients on the risks of developing uveitis before starting INREBIC therapy. Common uveitis symptoms include eye pain, redness, photophobia, floaters, and decreased vision. In case of symptoms, prompt ophthalmologic evaluation is recommended.
Major Adverse Cardiac Events (MACE)
Another Janus Kinase (JAK)-inhibitor has increased the risk of MACE, including cardiovascular death, myocardial infarction, and stroke (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which INREBIC is not indicated.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with INREBIC, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Another JAK-inhibitor has increased the risk of thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which INREBIC is not indicated. In patients with MF treated with INREBIC in clinical trials, the rates of thromboembolic events were similar in INREBIC and placebo treated patients.
Promptly evaluate and treat patients with symptoms of thrombosis.
Secondary Malignancies
Another JAK-inhibitor has increased the risk of lymphoma and other malignancies excluding nonmelanoma skin cancer (NMSC) (compared to those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which INREBIC is not indicated. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with INREBIC, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy, and patients who are current or past smokers.
Symptom Exacerbation Following Interruption or Discontinuation of Treatment
Following discontinuation of JAK-inhibitors, including INREBIC, signs and symptoms from myeloproliferative neoplasms may flare. Some patients with MF have experienced one or more of the following after discontinuing JAK-inhibitors: fever, respiratory distress, hypotension, disseminated intravascular coagulation, or multi-organ failure.
If one or more of these signs and symptoms occur after discontinuation of INREBIC, evaluate for and treat any intercurrent illness and consider restarting INREBIC. Instruct patients not to interrupt or discontinue therapy without consulting their healthcare provider. When discontinuing or interrupting therapy for reasons other thanpotentially life-threatening toxicities, consider tapering the dose of INREBIC gradually rather than discontinuing abruptly [see Dosage and Administration (2.2) ].
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Encephalopathy, including Wernicke's [see Warnings and Precautions (5.1) ]
- Anemia and Thrombocytopenia [see Warnings and Precautions (5.2) ]
- Gastrointestinal Toxicity [see Warnings and Precautions (5.3) ]
- Hepatic Toxicity [see Warnings and Precautions (5.4) ]
- Amylase and Lipase Elevation [see Warnings and Precautions (5.5) ]
- Uveitis [see Warnings and Precautions (5.6) ]
- Major Adverse Cardiac Events [see Warnings and Precautions (5.7) ]
- Thrombosis [see Warnings and Precautions (5.8) ]
- Secondary Malignancies [see Warnings and Precautions (5.9) ]
- Symptom Exacerbation Following Interruption or Discontinuation of Treatment [see Warnings and Precautions (5.10) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the WARNINGS AND PRECAUTIONS Section 5.1 Encephalopathy, including Wernicke's, reflect exposure to INREBIC as a single agent in 608 patients who received more than one dose (ranging from 30 mg to 800 mg) in Studies JAKARTA, ARD11936, JAKARTA2, ARD12042, ARD12888, TED12037/TED12015, INT12497, and TES13519, of whom 459 were patients with myelofibrosis, including 97 patients previously treated with ruxolitinib. Among the 608 patients receiving INREBIC, the median drug exposure was 37 weeks and the median number of cycles initiated was 9 cycles. Fifty-nine percent of 608 patients were exposed for 6 months or longer and 39% were exposed for 12 months or longer.
Using the dataset described above, the most common adverse reactions in >20% of patients (N=608) were diarrhea, nausea, anemia, vomiting, fatigue, thrombocytopenia, and constipation.
JAKARTA Trial
The safety of INREBIC was evaluated in the randomized treatment period of the JAKARTA trial [see Clinical Studies (14) ] . Key eligibility criteria included adult patients with intermediate-2 or high-risk primary MF or post-PV MF or post-ET MF with splenomegaly, platelet count ≥50 × 10 9 /L, and no splenectomy. Patients received INREBIC at 400 mg daily (n=96) or placebo (n=95). Among patients receiving INREBIC, 82% were exposed for more than 6 months and 65% for more than one year. Patients had a median duration of exposure to INREBIC 400 mg daily of 15.5 months compared with placebo where patients were treated for 6 months or until disease progression after which patients were allowed to crossover to active treatment. The median age of patients who received INREBIC was 65 years (range: 27 to 86 years), 59% were male, 90% were White, 8% were Asian, 1% were Black, 1% were Other, and 92% had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1.
Serious adverse reactions occurred in 21% of INREBIC-treated patients. Serious adverse reactions in ≥2% of patients receiving INREBIC 400 mg daily included cardiac failure (5%) and anemia (2%). Fatal adverse reactions of cardiogenic shock occurred in 1% of patients receiving INREBIC 400 mg daily.
Permanent discontinuation due to an adverse reaction occurred in 14% of patients receiving INREBIC. Most frequent reasons for permanent discontinuation in ≥2% of patients receiving INREBIC included cardiac failure (3%), thrombocytopenia, myocardial ischemia, diarrhea, and increased blood creatinine (2% each).
Dosage interruptions due to an adverse reaction during the randomized treatment period occurred in 21% of patients who received INREBIC. Adverse reactions requiring dosage interruption in >3% of patients who received INREBIC included diarrhea and nausea.
Dosage reductions due to an adverse reaction during the randomized treatment period occurred in 19% of patients who received INREBIC. Adverse reactions requiring dosage reduction in >2% of patients who received INREBIC included anemia (6%), diarrhea (3%), vomiting (3%), and thrombocytopenia (2%).
The most common adverse reactions (reported in ≥20%) were diarrhea, nausea, anemia, and vomiting.
Tables 3 and 4 summarize the common adverse reactions and laboratory abnormalities, respectively, in JAKARTA during randomized treatment.
| a CTCAE version 4.03. b Only 1 Grade 4 event (anemia). c Includes cystitis. | ||||
Adverse Reaction a | INREBIC 400 mg (n=96) | Placebo (n=95) | ||
All Grades % | Grade ≥3 b % | All Grades % | Grade ≥3 % | |
Diarrhea | 66 | 5 | 16 | 0 |
Nausea | 62 | 0 | 15 | 0 |
Anemia | 40 | 30 | 14 | 7 |
Vomiting | 39 | 3.1 | 5 | 0 |
Fatigue or asthenia | 19 | 5 | 16 | 1.1 |
Muscle spasms | 12 | 0 | 1.1 | 0 |
Blood creatinine increased | 10 | 1 | 1.1 | 0 |
Pain in extremity | 10 | 0 | 4.2 | 0 |
Alanine aminotransferase Increased | 9 | 0 | 1.1 | 0 |
Headache | 9 | 0 | 1.1 | 0 |
Weight increased | 9 | 0 | 4.2 | 0 |
Dizziness | 8 | 0 | 3.2 | 0 |
Bone pain | 8 | 0 | 2.1 | 0 |
Urinary tract infection c | 6 | 0 | 1.1 | 0 |
Dysuria | 6 | 0 | 0 | 0 |
Aspartate aminotransferase increased | 5 | 0 | 1.1 | 0 |
Clinically significant adverse reactions reported in 5% or less of patients: hypertension of all grades was reported in 4.2% of patients and Grade 3 or higher in 3% of INREBIC-treated patients.
Changes in selected postbaseline laboratory values that were observed are shown in Table 4 for the JAKARTA trial during randomized treatment.
INREBIC 400 mg (n=96) | Placebo (n=95) | |||
Laboratory Parameter | All Grades % | Grade ≥3 % | All Grades % | Grade ≥3 % |
Hematology | ||||
Anemia | 74 | 34 | 32 | 10 |
Thrombocytopenia | 47 | 12 | 26 | 10 |
Neutropenia | 23 | 5 | 13 | 3.3 |
Biochemistry | ||||
Creatinine increased | 59 | 3.1 | 19 | 1.1 |
ALT increased | 43 | 1 | 14 | 0 |
AST increased | 40 | 0 | 16 | 1.1 |
Lipase increased | 35 | 10 | 7 | 2.2 |
Hyponatremia | 26 | 5 | 11 | 4.3 |
Amylase increased | 24 | 2.1 | 5 | 0 |
In FEDR-MF-002, a randomized controlled post-marketing study of INREBIC (n=134) vs. best available therapy (BAT, n=67), the incidence of thiamine levels below the lower limit of normal (< 70 nmol/L) was 21% for INREBIC vs 5% for BAT. Thiamine levels < 30 nmol/L were not observed in the study. The median time to the first low thiamine level after initiation of INREBIC was 30 days. The frequency of low thiamine levels in participants receiving INREBIC was 2% in those receiving thiamine supplementation 100 mg orally per day vs. 39% in those not receiving thiamine supplementation.
Postmarketing Experience
The following adverse reactions have been identified during post approval use of INREBIC. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Eye Disorders: Uveitis
DRUG INTERACTIONS
- Strong CYP3A4 Inhibitors: Reduce INREBIC dose as recommended (2.3 , 7.1 ).
- Strong and Moderate CYP3A4 Inducers: Avoid use of INREBIC (7.1 ).
- CYP3A4, CYP2C19, or CYP2D6 Substrates: Dose modifications of substrates drugs may be needed (7.2 ).
- OCT2 and MATE1/2-K Substrates: Dose modifications of substrate drugs may be needed (7.2 ).
Effect of Other Drugs on INREBIC
Strong CYP3A4 Inhibitors
Coadministration of INREBIC with a strong CYP3A4 inhibitor increases fedratinib exposure [see Clinical Pharmacology (12.3) ] . Increased exposure may increase the risk of adverse reactions. Consider alternative therapies that do not strongly inhibit CYP3A4 activity. Alternatively, reduce the dose of INREBIC when administering with a strong CYP3A4 inhibitor [see Dosage and Administration (2.4) ] .
Strong and Moderate CYP3A4 Inducers
Coadministration of INREBIC with a strong or moderate CYP3A4 inducer can decrease fedratinib exposure [see Clinical Pharmacology (12.3) ] . Decreased exposure may reduce the effectiveness of INREBIC. Avoid INREBIC with strong and moderate CYP3A4 inducers.
Dual CYP3A4 and CYP2C19 Inhibitors
Coadministration of INREBIC with a dual CYP3A4 and CYP2C19 inhibitor increases fedratinib exposure [see Clinical Pharmacology (12.3) ] . Increased exposure may increase the risk of adverse reactions. Due to potential increase of exposure, patients taking concomitant dual CYP3A4 and CYP2C19 inhibitors require more intensive safety monitoring and, if necessary, dose modifications of INREBIC based on adverse reactions [see Dosage and Administration (2.6) ] .
Effect of INREBIC on Other Drugs
CYP3A4, CYP2C19, or CYP2D6 Substrate Drugs
Coadministration of INREBIC with drugs that are CYP3A4 substrates, CYP2C19 substrates, or CYP2D6 substrates increases the concentrations of these drugs, which may increase the risk of adverse reactions of these drugs [see Clinical Pharmacology (12.3) ] . Monitor for adverse reactions and adjust the dose of drugs that are CYP3A4, CYP2C19, or CYP2D6 substrates as necessary when coadministered with INREBIC.
OCT2 and MATE1/2-K Substrate Drugs
Coadministration of INREBIC with drugs that are renally excreted via organic cation transporter (OCT2) and multidrug and toxin extrusion (MATE)1/2-K can decrease renal clearance of those drugs [see Clinical Pharmacology (12.3) ] . Monitor for adverse reactions and consider dose modifications for drugs that are renally excreted via OCT2 or MATE1/2-K (e.g., metformin), as necessary when coadministered with INREBIC.
DESCRIPTION
INREBIC (fedratinib) is a kinase inhibitor with the chemical name N-tert-butyl-3-[(5-methyl-2-{[4-(2-pyrrolidin-1-ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide dihydrochloride monohydrate. Its empirical formula is C 27 H 36 N 6 O 3 S∙2HCl∙H 2 O and a molecular weight of 615.62. Fedratinib exhibits pH-dependent aqueous solubility; it is freely soluble in the acidic condition (>100 mg/mL at pH 1) and practically insoluble in the neutral condition (4 mcg/mL at pH 7.4). The chemical structure is:
INREBIC (fedratinib) is available as 100-mg (equivalent to 117.3 mg of fedratinib dihydrochloride monohydrate) hard gelatin capsules for oral administration. Each capsule contains inactive ingredients of silicified microcrystalline cellulose and sodium stearyl fumarate. The capsule shell contains gelatin, red iron oxide, titanium dioxide and white ink.
CLINICAL PHARMACOLOGY
Mechanism of Action
Fedratinib is an oral kinase inhibitor with activity against wild type and mutationally activated Janus Associated Kinase 2 (JAK2) and FMS-like tyrosine kinase 3 (FLT3). Fedratinib is a JAK2-selective inhibitor with higher inhibitory activity for JAK2 over family members JAK1, JAK3 and TYK2. Abnormal activation of JAK2 is associated with myeloproliferative neoplasms (MPNs), including myelofibrosis and polycythemia vera. In cell models expressing mutationally active JAK2 V617F or FLT3 ITD , fedratinib reduced phosphorylation of signal transducer and activator of transcription (STAT3/5) proteins, inhibited cell proliferation, and induced apoptotic cell death. In mouse models of JAK2 V617F -driven myeloproliferative disease, fedratinib blocked phosphorylation of STAT3/5, and improved survival, white blood cell counts, hematocrit, splenomegaly, and fibrosis.
Pharmacodynamics
Fedratinib inhibited cytokine-induced STAT3 phosphorylation in whole blood from patients with myelofibrosis. The inhibition of STAT3 phosphorylation was maximal approximately 2 hours after the first dose, with values returning to near baseline at 24 hours. After daily administration of fedratinib, levels of inhibition at steady state PK were similar to the maximal inhibition reached after the first dose of 300 (0.75 times the recommended dose), 400 or 500 mg (1.25 times the recommended dose) of fedratinib.
Cardiac Electrophysiology
The potential for QTc prolongation with fedratinib was evaluated in 31 patients with solid tumors. No large mean increase in the QTc interval (>20 ms) was detected with daily dosing of fedratinib 500 mg (1.25 times the recommended dose) for 14 days.
Pharmacokinetics
INREBIC at 300 mg to 500 mg once daily (0.75 to 1.25 times the recommended dose) results in a dose proportional increase in geometric mean fedratinib peak concentrations (C max ) and the area under the plasma concentration time curve over the dosing interval (AUC tau ). The mean steady state levels are achieved within 15 days of daily dosing. The mean accumulation ratio ranged between 3- to 4-fold.
At the dose of 400 mg once daily, the geometric mean (coefficient of variation, %CV) fedratinib C max is 1804 ng/mL (49%) and AUC tau is 26870 ng.hr/mL (43%) in patients with myelofibrosis.
Absorption
Following 400 mg once daily, fedratinib median time to peak concentrations (T max ) at steady-state is 3 hours (range: 2 to 4 hours).
Following the administration of a single 400 mg dose of fedratinib delivered orally or via nasogastric tube with contents from four 100-mg capsules dispersed in approximately 180 mL nutritional supplement (Ensure ® Plus), the systemic exposures of fedratinib are similar.
Effect of Food
A low-fat, low-calorie (total 162 calories: 6% from fat, 78% from carbohydrate and 16% from protein) or a high-fat, high-calorie (total 815 calories: 52% from fat, 33% from carbohydrate and 15% from protein) meal increased area under the curve over time to infinity (AUC inf ) up to 24% and C max up to 14% of a single 500 mg dose (1.25 times the recommended dose) of fedratinib.
Distribution
The apparent volume of distribution of fedratinib at steady-state is 1770 L in patients with myelofibrosis at 400 mg once daily dose. Fedratinib is 92% or greater bound to human plasma proteins.
Elimination
Fedratinib pharmacokinetics is characterized by a biphasic disposition with an effective half-life of 41 hours, a terminal half-life of approximately 114 hours, and apparent clearance (CL/F) (%CV) of 13 L/hr (51%) in patients with myelofibrosis.
Metabolism
Fedratinib is metabolized by CYP3A4, CYP2C19, and flavin-containing monooxygenase 3 (FMO3). Fedratinib accounts for approximately 80% of total circulating drug in plasma after oral administration.
Excretion
Following a single oral dose of radiolabeled fedratinib, 77% (23% unchanged) of the administered dose was excreted in feces and 5% (3% unchanged) was eliminated in urine.
Specific Populations
Age (20 years to 95 years), race (White, Asians), sex, body weight (40 kg to 135 kg), mild (Child-Pugh A), moderate (Child-Pugh B), or severe (Child-Pugh C) hepatic impairment, and mild (CLcr 60 mL/min to 89 mL/min by C-G) renal impairment did not have clinically meaningful effects on the pharmacokinetics of fedratinib.
Patients with Renal Impairment
Following a single 300 mg dose (0.75 times the recommended dose) of INREBIC, the AUC inf of fedratinib increased by 1.5-fold in subjects with moderate (CLcr 30 mL/min to 59 mL/min by C-G) renal impairment and 1.9-fold in subjects with severe (CLcr 15 mL/min to 29 mL/min by C-G) renal impairment, compared to that in subjects with normal renal function (CLcr ≥90 mL/min by C-G).
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effect of Strong and Moderate CYP3A4 Inhibitors
Coadministration of ketoconazole (strong CYP3A4 inhibitor: 200 mg twice daily) with a single dose of INREBIC (300 mg; 0.75 times the recommended dose) increased fedratinib AUC inf by 3-fold.
Coadministration of ketoconazole (400 mg once daily) with INREBIC 400 mg once daily is predicted to increase fedratinib AUC at steady state by 2-fold.
Coadministration of moderate CYP3A4 inhibitors, erythromycin (500 mg three times daily) or diltiazem (120 mg twice daily), with INREBIC 400 mg once daily is predicted to increase fedratinib AUC at steady state by 1.2-, and 1.1-fold, respectively.
Effect of Dual CYP3A4 and CYP2C19 Inhibitor
Coadministration of fluconazole (dual CYP3A4 and CYP2C19 inhibitor; 200 mg once daily) with a single dose of fedratinib (100 mg; 0.25 times the recommended dose) increased AUC inf by 1.7-fold.
Coadministration of fluconazole (200 mg once daily) with INREBIC 400 mg once daily is predicted to increase fedratinib AUC at steady state by approximately 1.5-fold.
Effect of Strong and Moderate CYP3A4 Inducers
Coadministration of rifampin (strong CYP3A4 inducer: 600 mg once daily) or efavirenz (moderate CYP3A4 inducer: 600 mg once daily) with a single dose of fedratinib (500 mg; 1.25 times the recommended dose) decreased AUC inf of fedratinib by approximately 81% or 47%, respectively.
Effect of Gastric Acid Reducing Agents
Coadministration of pantoprazole (proton pump inhibitor: 40 mg once daily) with a single dose of INREBIC (500 mg; 1.25 times the recommended dose) increased fedratinib AUC inf by 1.2-fold.
Effect of Fedratinib on Drugs that are CYP3A, CYP2C19, or CYP2D6 Substrates
Coadministration of a single dose of midazolam (CYP3A substrate: 2 mg), omeprazole (CYP2C19 substrate: 20 mg), and metoprolol (CYP2D6 substrate: 100 mg) with fedratinib increased midazolam, omeprazole, or metoprolol AUC inf by 4-, 3-, and 2-fold, respectively.
In Vitro and Clinical Transporter Studies
Fedratinib as a Substrate for Transporters :
Fedratinib is a substrate of P-glycoprotein (P-gp) but not breast cancer resistance protein (BCRP), BSEP, multidrug resistance protein (MRP2), and organic anion transporting polypeptide (OATP)1B1 and OATP1B3 in vitro .
Effect of Fedratinib on Transporter Substrates
Fedratinib inhibits P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, MATE1, and MATE2-K, but not BSEP, MRP2, and organic anion transporter (OAT)1 and OAT3 in vitro.
Coadministration of a single dose of fedratinib (600 mg; 1.5 times the recommended dose) with a single dose of digoxin (P-gp substrate: 0.25 mg), rosuvastatin (OATP1B1/1B3 and BCRP substrate: 10 mg), and metformin (OCT2 and MATE1/2-K substrate: 1000 mg) had no clinically meaningful effect on the AUC inf of digoxin, rosuvastatin, and metformin. Renal clearance of metformin was decreased by 36% in the presence of fedratinib. Contrary to unchanged PK, the glucose lowering PD effect of metformin in the presence of fedratinib appears reduced, with baseline adjusted glucose AUC being approximately 50% higher in response to an oral glucose challenge.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Fedratinib was not carcinogenic in the 6-month Tg.rasH2 transgenic mouse model.
Fedratinib was not mutagenic in a bacterial mutagenicity assay (Ames test) or clastogenic in in vitro chromosomal aberration assay (Chinese hamster ovary cells) or in vivo in a micronucleus test in rats.
In a fertility study in rats, fedratinib was administered for at least 70 days (males) and 14 days (females) prior to cohabitation and up to the implantation day (gestation day 7). Fedratinib had no effect on the estrous cycle parameters, mating performance, fertility, pregnancy rate or reproductive parameters in male or female rats at doses up to 30 mg/kg. The exposure (AUC) at the dose of 30 mg/kg/day is approximately 0.10 to 0.13 times the clinical exposure at the recommended daily dose.
Animal Toxicology and/or Pharmacology
The JAK/STAT pathway has been implicated in bone formation and metabolism, and its inhibition may cause bone abnormalities, e.g., in developing bone. There is currently no evidence of bone abnormalities in patients who received INREBIC.
CLINICAL STUDIES
JAKARTA
JAKARTA (NCT01437787) was a double-blind, randomized, placebo-controlled trial in patients with intermediate-2 or high-risk myelofibrosis, post-polycythemia vera myelofibrosis or post-essential thrombocythemia myelofibrosis with splenomegaly. A total of 289 patients were randomized to receive either INREBIC 500 mg (N=97), 400 mg (n=96) or placebo (n=96) once daily for at least 6 cycles. The median age was 65 years (range 27 to 86 years), 47% of patients were older than 65 years and 59% were male. Sixty-four percent (64%) of patients had primary MF, 26% had post-polycythemia vera MF, and 10% had post-essential thrombocythemia MF. Fifty-two percent (52%) of patients had intermediate-2 risk, and 48% had high-risk disease. The median baseline hemoglobin level was 10.2 g/dL. The median baseline platelet count was 214 × 10 9 /L; 16% of patients had a platelet count <100 × 10 9 /L and 84% of patients had a platelet count ≥100 × 10 9 /L. Patients had a baseline median palpable spleen length of 15 cm. Patients had a baseline median spleen volume as measured by magnetic resonance imaging (MRI) or computed tomography (CT) of 2568 mL (range of 316 to 8244 mL) (the upper limit of normal is approximately 300 mL). Patients underwent MRI or CT spleen volume assessment (after the third and sixth cycle) with a follow-up scan 4 weeks after Cycle 6.
The efficacy of INREBIC in the treatment of patients with primary or secondary myelofibrosis was established based upon the proportion of patients achieving greater than or equal to a 35% reduction from baseline in spleen volume at the End of Cycle 6 as measured by MRI or CT with a follow-up scan 4 weeks later.
Efficacy analyses are presented in Table 5.
Spleen Response by MRI/CT at the End of Cycle 6 with a Follow-up Scan 4 Weeks Later | INREBIC 400 mg N=96 n (%) | Placebo N=96 n (%) |
Number (%) of Patients with Spleen Volume Reduction by 35% or More | 35 (37) | 1 (1) |
p-value | p<0.0001 | |
Figure 1 shows the percent change in spleen volume from baseline for patients who have an evaluable MRI/CT at the End of Cycle 6.
Figure 1: Percent Change in Spleen Volume from Baseline at the End of Cycle 6 for Each Patient in the Phase 3 Study, JAKARTA
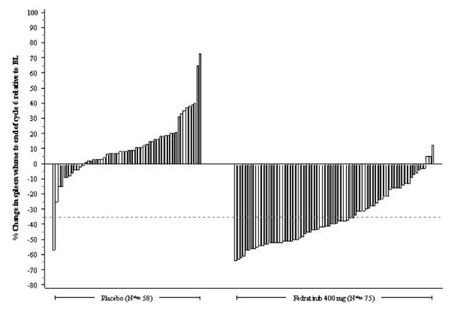
N•: Subjects with available percent change in spleen volume at EOC6.
Based on Kaplan-Meier estimates, the median duration of spleen response was 18.2 months for the INREBIC 400 mg group.
Additional outcomes included the proportion of patients with a 50% or greater reduction in Total Symptom Score from baseline to the End of Cycle 6 as measured by the modified Myelofibrosis Symptom Assessment Form (MFSAF) v2.0 diary.
The modified MFSAF v2.0 is a patient diary capturing the 6 core symptoms of MF: night sweats, itching, abdominal discomfort, early satiety, pain under ribs on left side, and bone or muscle pain. The modified MFSAF diary was completed daily during the week prior to Day 1 of each treatment cycle, and at the End of Cycle 6. Symptom scores ranged from 0 ("absent") to 10 ("worst imaginable"). These scores were added to create the Total Symptom Score, which has a maximum score of 60. At baseline, the mean Total Symptom Score was 17.95 in the 400 mg group and 15.45 in the placebo group.
The proportion of patients with a 50% or greater reduction in Total Symptom Score was 40% in the INREBIC 400 mg group and 9% in the placebo group (Table 6). Results are excluded for 22 patients: 6 patients with a baseline Total Symptom Score of zero (2 in the INREBIC 400 mg group and 4 in the placebo group) and 16 patients with missing baseline (5 in the INREBIC 400 mg group and 11 in the placebo group).
INREBIC 400 mg (N=89) n (%) | Placebo (N=81) n (%) | |
Number (%) of Patients with 50% or Greater Reduction in Total Symptom Score at the End of Cycle 6 | 36 (40) | 7 (9) |
p-value | p<0.0001 | |
Figure 2 shows the percent change in Total Symptom Score from baseline at the End of Cycle 6 for each patient.
Figure 2: Percent Change from Baseline in Total Symptom Score at the End of Cycle 6 for Each Patient in the Phase 3 Study, JAKARTA
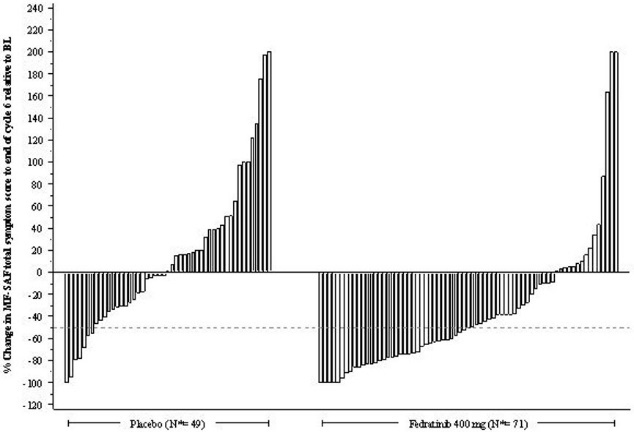
N•: Subjects with available percent change in Total Symptom Score at EOC6.
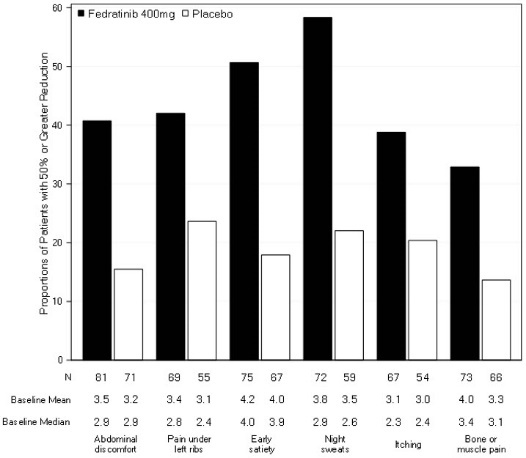
Figure 3 displays the proportion of patients with at least a 50% improvement in each of the individual symptoms that comprised the Total Symptom Score indicating that all 6 of the symptoms contributed to the higher Total Symptom Score response rate in the group treated with INREBIC.
Figure 3: Proportion of Patients Achieving 50% or Greater Reduction in Individual Symptom Scores at the End of Cycle 6 with Nonzero Baseline Scores
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
INREBIC (fedratinib) 100 mg capsules: Reddish brown, opaque, size 0 capsule, printed with "FEDR 100 mg" in white ink.
- Bottles of 120 capsules (NDC 59572-720-12)
Storage
Store below 30°C (86°F).
Mechanism of Action
Fedratinib is an oral kinase inhibitor with activity against wild type and mutationally activated Janus Associated Kinase 2 (JAK2) and FMS-like tyrosine kinase 3 (FLT3). Fedratinib is a JAK2-selective inhibitor with higher inhibitory activity for JAK2 over family members JAK1, JAK3 and TYK2. Abnormal activation of JAK2 is associated with myeloproliferative neoplasms (MPNs), including myelofibrosis and polycythemia vera. In cell models expressing mutationally active JAK2 V617F or FLT3 ITD , fedratinib reduced phosphorylation of signal transducer and activator of transcription (STAT3/5) proteins, inhibited cell proliferation, and induced apoptotic cell death. In mouse models of JAK2 V617F -driven myeloproliferative disease, fedratinib blocked phosphorylation of STAT3/5, and improved survival, white blood cell counts, hematocrit, splenomegaly, and fibrosis.