Get your patient on Miplyffa (Arimoclomol Citrate)
Patient education
Patient education materials
Patient support program
Dosing resources
Clinical information
Insurance resources
Prior authorization & coverage support
Other resources
Dosage & administration

Miplyffa prescribing information
INDICATIONS AND USAGE
MIPLYFFA is indicated for use in combination with miglustat for the treatment of neurological manifestations of Niemann-Pick disease type C (NPC) in adult and pediatric patients 2 years of age and older.
DOSAGE AND ADMINISTRATION
- Recommended MIPLYFFA oral dosage, in combination with miglustat, for patients with actual body weight of (2.1 ):
- 8 kg to 15 kg, is 47 mg three times a day
- > 15 kg to 30 kg, is 62 mg three times a day
- > 30 kg to 55 kg, is 93 mg three times a day
- > 55 kg, is 124 mg three times a day
- Administer with or without food. (2.1 )
- See full prescribing information for recommended dosage in patients with an eGFR ≥ 15 to < 50 mL/minute. (2.2 )
- See full prescribing information for instructions on preparation and administration. (2.3 )
Recommended Dosage
The recommended oral dosage of MIPLYFFA, in combination with miglustat, for patients with an actual body weight of:
- 8 kg to 15 kg, is 47 mg three times a day
- > 15 kg to 30 kg, is 62 mg three times a day
- > 30 kg to 55 kg, is 93 mg three times a day
- > 55 kg, is 124 mg three times a day
Administer MIPLYFFA with or without food.
Missed Dose
If a dose of MIPLYFFA is missed, advise the patient to skip the missed dose and to resume taking the prescribed dose at the next scheduled time.
Recommended Dosage in Patients with Renal Impairment
The recommended dosage of MIPLYFFA, in combination with miglustat, in patients with an eGFR ≥ 50 mL/minute is the same as the recommended dosage in patients with normal renal function [see Dosage and Administration (2.1 )].
For patients with an eGFR ≥ 15 to < 50 mL/minute, the recommended oral dosage of MIPLYFFA, in combination with miglustat, for patients with an actual body weight of [see Use in Specific Populations (8.6 ) and Clinical Pharmacology (12.3 )] :
- 8 kg to 15 kg, is 47 mg two times a day
- > 15 kg to 30 kg, is 62 mg two times a day
- > 30 kg to 55 kg, is 93 mg two times a day
- > 55 kg, is 124 mg two times a day
Administer MIPLYFFA with or without food.
Preparation and Administration Instructions
Swallow MIPLYFFA whole. However, for patients who have difficulty swallowing capsules, administer MIPLYFFA in one of two ways:
Oral Administration
- Carefully open the capsule and sprinkle the entire contents into 15 mL of water or apple juice or 15 mL of soft food (e.g., applesauce, pudding, or yogurt).
- Stir the mixture for 15 seconds.
- Consume the entire mixture immediately.
Feeding Tube Administration (nasogastric or gastric tube)
- Carefully open the capsule and sprinkle the entire contents into 20 mL of water. Do not add the capsule contents to other liquids besides water.
- Stir the mixture for 15 seconds.
- Administer the entire mixture immediately via feeding tube.
- Flush the feeding tube with 5 mL of water after administration.
Do not save the mixture for later use.
DOSAGE FORMS AND STRENGTHS
MIPLYFFA (arimoclomol) capsules are available as follows:
| MIPLYFFA Strength | Description of Capsules |
| 47 mg | White opaque body with black printing “47”, and green opaque cap with black printing “OZ” |
| 62 mg | White opaque body with black printing “62”, and yellow opaque cap with black printing “OZ” |
| 93 mg | White opaque body with black printing “93”, and orange opaque cap with black printing “OZ” |
| 124 mg | White opaque body with black printing “124”, and red opaque cap with black printing “OZ” |
USE IN SPECIFIC POPULATIONS
- Females and Males of Reproductive Potential: Based on animal findings, MIPLYFFA may impair fertility. (8.3 )
Pregnancy
Risk Summary
Based on findings from animal reproduction studies, MIPLYFFA may cause embryofetal harm when administered during pregnancy. There are no available data on MIPLYFFA use in pregnant females to evaluate a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Advise pregnant females of the potential risk to the fetus.
In animal reproduction studies, oral administration of arimoclomol to pregnant rats and rabbits during organogenesis resulted in post-implantation loss and structural abnormalities in offspring. These occurred at exposures equal to or greater than 10- and 5-fold, in rats and rabbits respectively, the human exposure at the maximum recommended human daily dose of 372 mg ( see Data ).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data: In an embryofetal development study in pregnant rats, once daily oral doses of arimoclomol were administered throughout organogenesis from gestation day 6 to 17. Increased post-implantation loss was observed at 10-fold the human exposure, based on AUC at the MRHDD, together with increased ossification in the vertebrae and dilated brain ventricles in litters of dams dosed at equal to or greater than 8-fold the human exposure at the MRHDD.
In another embryofetal development study in pregnant rats in which arimoclomol was administered three times-daily throughout organogenesis from gestation day 6 to 17, there was an increase in postimplantation loss and reduced maternal, placental, and fetal weights at 14-fold the human exposure, based on AUC at the MRHD. In addition, fetuses of dams treated at this exposure level exhibited domed craniums, partially split sternum, hydrocephaly, dilated brain ventricles, dilated interventricular foramen, misaligned and misshapen hemicentres and misaligned ossification sites in the sternebrae, misaligned costal cartilage, increased ossification, cerebral and cerebellar hemorrhages, bipartite supraoccipital, large interparietal bone, marked enlargement of the anterior and posterior fontanelles, hyoid bone, meningocele and fusion of the jugal and maxilla.
In an embryofetal development study in pregnant rabbits, arimoclomol was administered once daily by oral gavage throughout organogenesis from gestation day 7 to 19. Increased incidences of minor skeletal variations (bent hyoid and unossified phalanx) were observed at 5-fold the human AUC at the MRHDD, coinciding with an adverse reduction of maternal body weight.
In a pre- and postnatal development study in pregnant rats, oral arimoclomol was administered from gestation day 6 to lactation day 21. Increased embryofetal lethality and reduced pup weight, with a slight reduction in maternal body weight gain, were observed at 10-fold the human AUC at the MRHDD.
Lactation
Risk Summary
There are no data on the presence of arimoclomol in human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for MIPLYFFA and any potential adverse effects on the breastfed infant from MIPLYFFA or from the underlying maternal condition.
Females and Males of Reproductive Potential
MIPLYFFA may cause embryofetal harm when administered to a pregnant female [see Use in Specific Populations (8.1 )] .
Contraception
Females: Consider pregnancy planning and prevention for females of reproductive potential.
Infertility
Based on findings from animal studies, MIPLYFFA may impair fertility in females and males of reproductive potential. In a rat fertility study, oral administration of arimoclomol resulted in decreased male and female fertility at 9-fold and increased pre-implantation loss at 5-fold the human exposure, based on AUC at MRHD. It is not known if these effects are reversible [see Nonclinical Toxicology (13.1 )] .
Pediatric Use
The safety and effectiveness of MIPLYFFA in combination with miglustat for the treatment of neurological manifestations of NPC have been established in pediatric patients 2 years of age and older. Use of MIPLYFFA in combination with miglustat for this indication is supported by evidence from a randomized, double-blind, placebo-controlled 12-month trial (Trial 1) [see Clinical Studies (14 )].
The safety and effectiveness of MIPLYFFA have not been established in pediatric patients younger than 2 years of age.
Juvenile Animal Toxicity Data
In juvenile toxicity studies in rats, increased incidences of renal pelvic dilatation were observed at all dose levels corresponding to 4-, 7- and 17-fold the human exposure based on AUC at MRHDD after both 2 and 8 weeks of dosing when animals were dosed from postnatal day 7.
Geriatric Use
NPC is largely a disease of pediatric and young adult patients. Clinical trials of MIPLYFFA in combination with miglustat in patients with NPC did not include patients aged 65 years or older.
Renal Impairment
The recommended MIPLYFFA dosage in combination with miglustat in patients with an eGFR 15 mL/minute to < 50 mL/minute is lower than the recommended dosage (less frequent dosing) in patients with normal renal function [see Dosage and Administration (2.2 )]. The recommended dosage of MIPLYFFA in combination with miglustat in patients with an eGFR ≥ 50 mL/minute is the same as the recommended dosage in patients with normal renal function.
Plasma concentrations of arimoclomol increased in patients with eGFR ≥ 15 mL/minute to < 50 mL/minute [see Clinical Pharmacology (12.3 )].
The pharmacokinetics of arimoclomol have not been evaluated in patients with eGFR < 15 mL/minute.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions : Urticaria and angioedema have been reported. Discontinue MIPLYFFA in patients who develop these adverse reactions. (5.1 )
- Embryofetal Toxicity : May cause fetal harm. Advise pregnant females of the potential risk to the fetus and consider pregnancy planning and prevention. (5.2 )
- Increased Creatinine without Affecting Glomerular Function: Mean increases in serum creatinine of 10-20% have been reported. Use alternative measures to assess renal function which are not based on creatinine. (5.3 )
Hypersensitivity Reactions
Hypersensitivity reactions such as urticaria and angioedema have been reported in patients treated with MIPLYFFA during Trial 1 [see Clinical Studies (14 )] : two patients reported both urticaria and angioedema (6%) and one patient (3%) experienced urticaria alone. The reactions occurred within the first two months of treatment. Discontinue MIPLYFFA in patients who develop severe hypersensitivity reactions. If a mild or moderate hypersensitivity reaction occurs, stop MIPLYFFA and treat promptly. Monitor the patient until signs and symptoms resolve [see Adverse Reactions (6.1 )].
Embryofetal Toxicity
Based on findings from animal reproduction studies, MIPLYFFA may cause embryofetal harm when administered during pregnancy. In animal reproduction studies, oral administration of arimoclomol to pregnant rats and rabbits resulted in post-implantation loss and structural abnormalities in offspring. These occurred at exposures equal to or greater than 10- and 5-fold, for rats and rabbits respectively, the human exposure at the maximum recommended human daily dose of 372 mg. Advise pregnant females of the potential risk to the fetus. Consider pregnancy planning and prevention for females of reproductive potential [see Use in Specific Populations (8.1 , 8.3 )] .
Increased Creatinine without Affecting Glomerular Function
Across clinical trials of MIPLYFFA consisting of patients with NPC, healthy subjects, and patients with other diseases, there were mean increases in serum creatinine of 10% to 20% compared to baseline. These increases occurred mostly in the first month of MIPLYFFA treatment and were not associated with changes in glomerular function. The increases in serum creatinine may be due to inhibition of renal tubular secretion transporters [see Drug Interactions (7 ) and Clinical Pharmacology (12.2 , 12.3 )].
During MIPLYFFA treatment, use alternative measures that are not based on creatinine to assess renal function such as BUN, cystatin C, or measured GFR. Increases in creatinine reversed upon MIPLYFFA discontinuation [see Clinical Pharmacology (12.2 )] .
ADVERSE REACTIONS
The following clinically significant adverse reactions are described below and elsewhere in the labeling:
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of MIPLYFFA was evaluated in a randomized, double-blind, placebo-controlled, 12-month trial (Trial 1), which included 50 patients 2 to 19 years old with NPC [see Clinical Studies (14 )] . Patients received weight-adjusted doses of MIPLYFFA (31 to 124 mg orally three times daily); 28 of the patients were exposed to MIPLYFFA for one year. In Trial 1, 78% of patients received miglustat. Forty-one out of 50 patients that were enrolled in Trial 1 continued into an open-label extension trial, which included 39 patients treated with MIPLYFFA for more than 1 year, 34 patients treated with MIPLYFFA for more than 2 years, and 17 patients treated with MIPLYFFA for more than 5 years.
The most common adverse reactions in Trial 1 (≥ 15%) in MIPLYFFA-treated patients who also received miglustat were upper respiratory tract infection, diarrhea, and decreased weight. Three (6%) of the MIPLYFFA-treated patients had the following adverse reactions that led to withdrawal from Trial 1: increased serum creatinine (one patient), and progressive urticaria and angioedema (two patients). Serious adverse reactions reported in MIPLYFFA-treated patients in Trial 1 were hypersensitivity reactions including urticaria and angioedema.
Table 1 shows common adverse reactions in Trial 1 that occurred in at least 8% of MIPLYFFA-treated patients who also received miglustat.
• Upper Respiratory Tract Infection: Combined incidence of upper respiratory tract infection and rhinitis. | ||
•• Urticaria: Includes one patient in which urticaria occurred alone (3%) and two patients who had urticaria with angioedema (6%) | ||
| Adverse Reaction | MIPLYFFA with miglustat N=26 n (%) | Placebo with miglustat N=13 n (%) |
| Upper Respiratory Tract Infection• | 8 (31) | 2 (15) |
| Diarrhea | 6 (23) | 3 (23) |
| Decreased Weight | 4 (15) | 0 |
| Decreased appetite | 3 (12) | 0 |
| Tremor | 3 (12) | 0 |
| Urticaria•• | 3 (12) | 0 |
| Headache | 3 (12) | 1 (8) |
| Lower respiratory tract infection | 3 (12) | 1 (8) |
| Seizure | 3 (12) | 1 (8) |
Decreased Weight
Adverse reactions of decreased weight were observed in four patients, who were also receiving concomitant miglustat during the trial. The decrease in weight resolved in all but one of the patients. The mean duration of the weight decrease was 33 days and ranged from 22 to 60 days. One patient had two separate instances of weight loss during the trial, lasting 22 and 24 days respectively. The mean weight loss was approximately 6% from baseline in all patients and MIPLYFFA administration was not interrupted in any patient.
Laboratory Findings
Thrombocytopenia: Thrombocytopenia was observed in three patients during the trial, all of whom were receiving miglustat for six months or longer at the time of enrollment. In two of these patients, the thrombocytopenia was present at baseline and persisted throughout the trial. In the other patient, the thrombocytopenia developed and resolved during the trial.
Increased Creatinine: Across clinical trials in patients with NPC, healthy subjects, and patients with other diseases, increases in serum creatinine (mean increase was 10-20%) occurred mainly within the first month of dosing and were reversible upon treatment discontinuation.
DRUG INTERACTIONS
- Substrates of the Organic Cationic Transporter 2 (OCT2 substrates) : Monitor for adverse reactions and reduce the dosage of the OCT2 substrate. (7.1 )
Effect of MIPLYFFA on Other Drugs
Arimoclomol is an inhibitor of the organic cationic transporter 2 (OCT2) transporter and may increase the exposure of drugs that are OCT2 substrates [see Clinical Pharmacology (12.3 )] . When MIPLYFFA is used concomitantly with OCT2 substrates, monitor for adverse reactions and reduce the dosage of the OCT2 substrate.
DESCRIPTION
MIPLYFFA capsules contain arimoclomol citrate. Arimoclomol citrate is a crystalline powder of white to off-white color that is freely soluble in water. The chemical name is N -[(2R, Z )-2-hydroxy-3-(1piperidyl)propoxy]pyridine-3-carboximidoyl chloride, 1-oxide, citrate.
The empirical formula is C 20 H 28 ClN 3 O 10 and the molecular weight is 505.90 g/mol. The chemical structure is:
MYPLIFFA contains 47 mg, 62 mg, 93 mg, or 124 mg of arimoclomol (equivalent to 75 mg, 100 mg, 150 mg, or 200 mg of arimoclomol citrate). The inactive ingredients are microcrystalline cellulose (MCC) and magnesium stearate. The inactive ingredients are not water soluble and will remain undissolved if the contents of the capsule are added to beverages or soft food [see Dosage and Administration (2.3 )].
The capsule shells contain hypromellose, titanium dioxide, and one or more of the following: Brilliant Blue FCF-FD&C Blue 1, Yellow iron oxide and Red iron oxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
The mechanism(s) by which arimoclomol exerts its clinical effects in patients with NPC is unknown.
Pharmacodynamics
The effect of arimoclomol (744 mg/day for 28 days) on serum creatinine was assessed in 16 healthy male subjects. A reversible increase in mean serum creatinine of 19% was observed after 21 days of treatment. No effect on glomerular function (GFR, 125 I-iothalamate clearance) or renal hemodynamics (effective renal plasma flow (ERPF); 131 I-hippuran clearance) was observed [see Warnings and Precautions (5.3 ), Adverse Reactions (6.1 ), and Drug Interactions (7 )].
Cardiac Electrophysiology
The effect of arimoclomol 124 mg and 372 mg (3 times the maximum recommended dosage) administered three times a day (372 mg/day and 1116 mg/day) on ECG was evaluated in a randomized, partially double-blind, placebo-, and positive-controlled (moxifloxacin 400 mg), multipledose, 4-way crossover trial in 34 healthy male subjects. In this trial, arimoclomol did not prolong the QTc interval to a clinically relevant extent.
Pharmacokinetics
Arimoclomol exhibited linear and dose proportional pharmacokinetics following oral administration of doses ranging from 62 to 372 mg (three times the maximum recommended dosage) three times a day in healthy subjects. The plasma C max and AUC 0-8hr of arimoclomol at the 248 mg dose (two times the maximum recommended dosage) are summarized in Table 2 .
| Plasma PK Parameters | 248 mg arimoclomol oral administration three times a day | |
| Day 1 (first dose) | Day 6 (steady state) | |
| AUC 0-8hr (hr·ng/mL) | 5317 (17%) | 7207 (19%) |
| C max (ng/mL) | 1749 (49%) | 2090 (23%) |
Absorption
The absolute bioavailability of arimoclomol following oral administration has not been determined. The median time to reach maximum plasma arimoclomol concentration (t max ) was approximately 0.5 hours.
Effect of Food
No clinically significant difference in arimoclomol pharmacokinetics were observed following administration of a high-fat meal (1000 calories, 60% fat) to healthy subjects.
Distribution
The mean apparent volume of distribution (V Z /F) of arimoclomol at steady state in healthy adult subjects is 211 L. A dose-dependent increase in arimoclomol cerebral spinal fluid concentrations was seen at steady state. Plasma protein binding of arimoclomol is approximately 10%.
Elimination
The elimination half-life of arimoclomol is approximately 4 hours. The mean apparent clearance of arimoclomol (CL/F) at steady state is 34 L/hr in healthy adult subjects.
Metabolism: Arimoclomol is predominantly metabolized through glutathionation, O -glucuronidation and NO-oxime cleavage.
Excretion: Following a single dose of radiolabeled arimoclomol 100 mg to healthy male subjects under fasted conditions, approximately 12% of the dose was recovered in feces and 77.5% in urine (42% unchanged).
Drug Interaction Studies
In Vitro Studies: Cytochrome P450 (CYP) Enzymes: Arimoclomol is not an inhibitor or inducer of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4/5.
Transporter Systems: Arimoclomol is an inhibitor of OCT2 and may cause relevant changes in exposure of OCT2 substrates including the endogenous substrate creatinine [see Drug Interactions (7.1 )]. Arimoclomol is not an inhibitor of P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, MATE1 and MATE2-K transporters. Arimoclomol is a substrate of MATE1 and MATE2-K transporters; MATE1 and MATE2-K inhibitors are not expected to have a clinically relevant effect on arimoclomol exposure.
Specific Populations
Pediatric Patients: In pediatric NPC patients who received the recommended dosing regimens, the estimated steady state mean ± SD serum C trough and C max arimoclomol concentrations are 206 ± 60 and 523 ± 194 ng/mL, respectively.
Patients with Renal Impairment: In a renal impairment study, subjects with moderate to severe renal impairment (estimated glomerular filtration rate [eGFR] 15-49 mL/minute) had an approximately 2-fold increase in total exposure to arimoclomol (AUC inf ) compared to subjects with normal renal function (eGFR ≥ 90 mL/minute) [see Dosage and Administration (2.2 )] . There was no clinically meaningful difference in exposure to arimoclomol in subjects with an eGFR of ≥ 50 mL/minute. MIPLYFFA was not evaluated in patients with eGFR < 15 mL/minute.
Patients with Hepatic Impairment: No clinically relevant differences in arimoclomol pharmacokinetics were observed in patients with mild to moderate hepatic impairment (Child-Pugh Score A or B) compared to subjects with normal hepatic function. MIPLYFFA has not been studied in patients with severe hepatic impairment (Child-Pugh Criteria C).
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 2-year carcinogenicity study in Han Wistar rats, and a 26-week carcinogenicity study in Transgenic rasH2 mice, oral administration of arimoclomol did not increase the incidence of tumors at systemic exposures that were approximately 8-fold and 11-fold the human exposure, based on AUC at the MRHD.
Mutagenesis
Arimoclomol was not mutagenic or clastogenic in a standard battery of genotoxicity tests (bacterial mutagenicity [Ames], chromosomal aberration in Chinese Hamster Ovary cells, mouse lymphoma forward mutation, mouse and rat bone marrow micronucleus).
Impairment of Fertility
In a fertility and early embryonic development study in rats, once daily oral arimoclomol doses were administered to males for 4 weeks prior to and throughout mating (for a total of 10 weeks) and to females for 2 weeks prior to mating and to gestation day 6. A reduction in fertility and fecundity indices was noted for both sexes at 9-fold the human exposure, based on AUC at the MRHD. At the same dose level, a reduction in the number of corpora lutea was observed in females, and reduced sperm motility, immotile sperm, reduced sperm count and increased sperm abnormalities were observed in males. A dose-dependent increase in preimplantation loss was noted across all treated groups (equal to or greater than 5-fold the human exposure, based on AUC at the MRHD), which resulted in a reduction in the number of live embryos [see Use in Specific Populations (8.3 )].
CLINICAL STUDIES
Safety and effectiveness of MIPLYFFA were assessed in Trial 1, a randomized, double-blind, placebocontrolled, 12-month trial in patients 2 to 19 years of age who had a molecularly confirmed diagnosis of NPC (NCT02612129). Fifty patients were randomized 2:1 to treatment with weight-adjusted MIPLYFFA (31 to 124 mg) or placebo orally three times per day. The randomization was stratified by miglustat use status at baseline.
Efficacy assessments, including the rescored 4-domain NPC Clinical Severity Scale (R4DNPCCSS) score, were performed at baseline and every 3 months until 12 months of treatment. The R4DNPCCSS is a measure of NPC disease progression that consists of the four items assessing ambulation, speech, swallow, and fine motor skills that patients with NPC and their caregivers and physicians have identified as most relevant with higher scores representing greater severity of disease.
In Trial 1, 76% and 81% of patients in the MIPLYFFA and placebo groups, respectively, received miglustat six months or longer prior to the time of enrollment. For the subgroup of patients who also received miglustat at enrollment, the mean age was 11.6 years, the mean time since first NPC symptom was 8.5 years, and the mean age at onset of first neurological symptom was 4.9 years. In this subgroup, 56% of patients were females, 87% were white, 5% were Asian, 3% were native Hawaiian or other Pacific Islander, and 5% were unknown. The mean baseline R4DNPCCSS score was higher in the MIPLYFFA group (n=26; mean=8.9) than the placebo group (n=13; mean=7), with an overall mean R4DNPCCSS score of 8.3.
In the MIPLYFFA group, four patients discontinued the study: one patient due to consent withdrawal and three patients due to adverse reactions [see Adverse Reactions (6.1 )]. In the placebo group, one patient discontinued the study due to an adverse event.
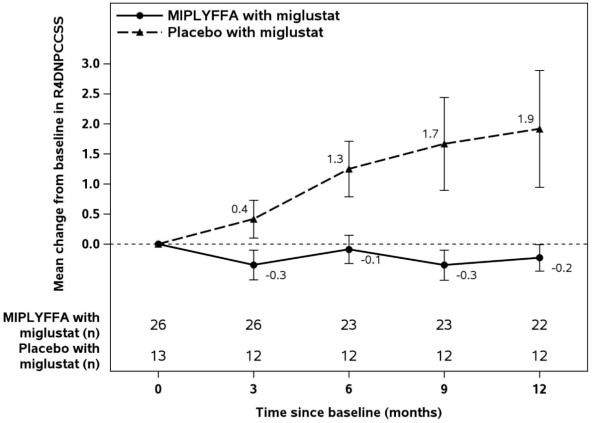
Table 3 displays the change from baseline in R4DNPCCSS score at month 12 in patients 2 to 19 years of age with NPC who also received miglustat in Trial 1. See Figure 1 for the mean change in R4DNPCCSS score from baseline over time by treatment group in patients 2 to 19 years of age with NPC who also received miglustat.
[1] Changes in R4DNPCCSS score from baseline to month 12 were compared using an analysis of variance (ANCOVA) model fitted with treatment and baseline R4DNPCCSS score as covariate. | ||||
| R4DNPCCSS Score | ||||
| Baseline | Change from Baseline to Month 12 | |||
| MIPLYFFA with miglustat (N=26) | Placebo with miglustat (N=13) | MIPLYFFA with miglustat (N=22) | Placebo with miglustat (N=12) | |
| Mean (SD) | 8.9 (6.1) | 7 (5.8) | -0.2 (1) | 1.9 (3.4) |
| Median | 7.5 | 5 | 0 | 1 |
| LS Mean (SE) | -0.2 (0.5) | 2 (0.7) | ||
| Placebo-subtracted Difference (95% CI) [1] | -2.2 (-3.8, -0.6) | |||
Figure 1: MIPLYFFA and Placebo Mean Change from Baseline (± SE) in R4DNPCCSS Score Over Time in Patients 2 to 19 Years of Age with NPC Who Also Received Miglustat (Trial 1)
There were insufficient data to determine the effectiveness of the use of MIPLYFFA without miglustat for the treatment of neurological manifestations in patients with NPC.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
MIPLYFFA (arimoclomol) capsules are supplied in high-density polyethylene (HDPE) bottles with child-resistant closure as follows:
| MIPLYFFA Strength | Capsules Description | Package Configuration | NDC No. |
| 47 mg | White opaque body with black printing “47”, and green opaque cap with black printing “OZ” | Bottle of 90 capsules | 72542-147-01 |
| 62 mg | White opaque body with black printing “62”, and yellow opaque cap with black printing “OZ”. | Bottle of 90 capsules | 72542-162-01 |
| 93 mg | White opaque body with black printing “93”, and orange opaque cap with black printing “OZ” | Bottle of 90 capsules | 72542-193-01 |
| 124 mg | White opaque body with black printing “124”, and red opaque cap with black printing “OZ” | Bottle of 90 capsules | 72542-124-01 |
Storage and Handling
Store MIPLYFFA at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Store in original container. Protect from light.
INSTRUCTIONS FOR USE
MIPLYFFA ® [mye plye' fah]
(arimoclomol) capsules for oral use
This Instructions for Use contains information on how to take or give MIPLYFFA. Read this Instructions for Use before taking or giving MIPLYFFA and each time you get a refill. There may be new information. This Instructions for Use does not take the place of talking to your or your child's healthcare provider about your or your child's medical condition or treatment.
Important Information You Need to Know Before Taking or Giving MIPLYFFA
- Take or give MIPLYFFA capsules exactly as instructed by your healthcare provider.
- Your healthcare provider will tell you the number of MIPLYFFA capsules to take or give for the prescribed dose.
- MIPLYFFA capsules can be swallowed whole with or without food.
- If you or your child have difficulty swallowing capsules, MIPLYFFA capsules can be opened and:
- can be taken or given by mouth ( see Step 1 to Step 5 ) or
- through a feeding tube ( see Step 6 to 13 ).
- If you miss a dose of MIPLYFFA, skip the missed dose and take or give the next dose of MIPLYFFA at your regularly scheduled time.
Preparing to take or give MIPLYFFA by mouth if you have difficulty swallowing capsules
Supplies needed to take or give 1 dose of MIPLYFFA:
- 1 MIPLYFFA capsule,
- 15 mL of liquid (water or apple juice) or soft food (apple sauce, yogurt, or pudding).
- small cup
- spoon
| Step 1: Add 15 mL of water, apple juice, apple sauce, yogurt, or pudding into a small cup. Adding MIPLYFFA to any other liquids is not recommended. | ||
| Step 2: Carefully open 1 MIPLYFFA capsule by twisting the top and bottom of the capsule (see Figure A ). Sprinkle the entire contents of the capsule into the small cup (see Figure B ). |  Figure A Figure A |  Figure B Figure B |
Step 3: Stir the mixture for 15 seconds with a spoon (see Figure C ).
| 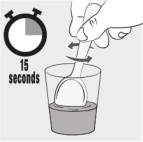 Figure C Figure C | |
| Step 4: Swallow all of the mixture right away (see Figure D ). Do not save any of the mixture for later use. Throw away the MIPLYFFA mixture if not swallowed right away. | 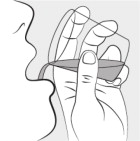 Figure D Figure D | |
| Step 5: Throw away the empty MIPLYFFA capsule shells in the household trash. | ||
Preparing to take or give MIPLYFFA through a feeding tube (nasogastric or gastric tube)
Supplies needed to take 1 dose of MIPLYFFA:
- 1 MIPLYFFA capsule
- 1 feeding tube syringe (at least 20 mL)
- water
- small cup
- spoon
| Step 6: Use the feeding tube syringe to measure 20 mL of water into a small cup (see Figure E ). | 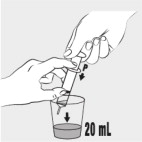 Figure E Figure E | |
| Step 7: Carefully open 1 MIPLYFFA capsule by twisting the top and bottom of capsule (see Figure F ). Sprinkle the entire contents of the capsule into the small cup (see Figure G ). Do not add the contents of the capsule to any other liquids besides water. | 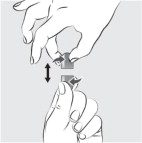 Figure F Figure F | 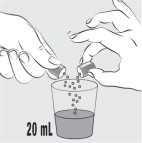 Figure G Figure G |
| Step 8: Stir the mixture for 15 seconds with a spoon (see Figure H ). • After mixing, some of the capsule contents may remain in the mixture. | 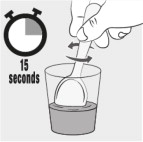 Figure H Figure H | |
| Step 9: Place the feeding tube syringe in the mixture. Pull up on the feeding tube syringe plunger (indicated with a “ P ” on the figure) to withdraw the whole mixture into the feeding tube syringe (see Figure I ). | 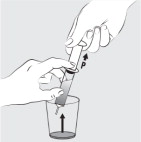 Figure I Figure I | |
| Step 10: Attach the feeding tube syringe to the feeding tube. Slowly push the plunger ( P ) to give or take the mixture right away through the feeding tube (see Figure J ). Do not save any of the mixture for later use. Throw away the MIPLYFFA mixture if not given or taken right away. |  Figure J Figure J | |
| Step 11: Disconnect the empty feeding tube syringe from the feeding tube. Fill the small cup with water. | ||
| Step 12: Fill the syringe with 5 mL of water and slowly push the plunger ( P ) to flush the feeding tube with water (see Figure K ). | 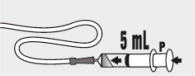 Figure K Figure K | |
| Step 13: Throw away the empty MIPLYFFA capsule shells in the household trash. | ||
Storing MIPLYFFA
- Store MIPLYFFA at room temperature between 68°F to 77°F (20°C to 25°C).
- Store in original container. Protect from light.
Manufactured for: Zevra Therapeutics, Inc. Boston, MA 02110, USA © 2024, 2025 Zevra Therapeutics, Inc. All rights reserved. MIPLYFFA is a registered trademark of Zevra Therapeutics, Inc.
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: March/2026
Mechanism of Action
The mechanism(s) by which arimoclomol exerts its clinical effects in patients with NPC is unknown.