Get your patient on Rezdiffra (Resmetirom)
Patient education
Patient education materials
Patient support program
Dosing resources
Clinical information
Insurance resources
Prior authorization & coverage support
Financial assistance & copay programs
Other resources
Dosage & administration

Rezdiffra prescribing information
INDICATIONS AND USAGE
REZDIFFRA is indicated in conjunction with diet and exercise for the treatment of adults with noncirrhotic nonalcoholic steatohepatitis (NASH) with moderate to advanced liver fibrosis (consistent with stages F2 to F3 fibrosis).
This indication is approved under accelerated approval based on improvement of NASH and fibrosis [see Clinical Studies (14) ] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Limitations of Use
Avoid use of REZDIFFRA in patients with decompensated cirrhosis [see Use in Specific Populations (8.7) , Clinical Pharmacology (12.3) ] .
DOSAGE AND ADMINISTRATION
Recommended Dosage and Administration
The recommended dosage of REZDIFFRA is based on actual body weight. For patients weighing:
- <100 kg, the recommended dosage is 80 mg orally once daily.
- ≥100 kg, the recommended dosage is 100 mg orally once daily.
Administer REZDIFFRA with or without food [see Clinical Pharmacology (12.3) ] .
Swallow REZDIFFRA tablets whole; do not split, crush, or chew tablets.
Advise patients that if a dose is missed, do not take the missed dose and resume with the next scheduled dose.
Dosage Modifications for CYP2C8 Inhibitors
Concomitant use of REZDIFFRA with strong CYP2C8 inhibitors (e.g., gemfibrozil) is not recommended [see Drug Interactions (7.1) ] .
If REZDIFFRA is used concomitantly with a moderate CYP2C8 inhibitor (e.g., clopidogrel) [see Drug Interactions (7.1) ] , reduce the dosage of REZDIFFRA:
- <100 kg, reduce the dosage of REZDIFFRA to 60 mg once daily.
- ≥100 kg, reduce the dosage of REZDIFFRA to 80 mg once daily.
DOSAGE FORMS AND STRENGTHS
REZDIFFRA Tablets:
- 60 mg: white oval-shaped film-coated tablets debossed with “P60” on one side and plain on the other side.
- 80 mg: yellow, oval-shaped, film-coated tablets debossed with “P80” on one side and plain on the other side.
- 100 mg: beige to pink, oval-shaped, film-coated tablets debossed with “P100” on one side and plain on the other side.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
There are no available data on REZDIFFRA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. There are risks to the mother and fetus related to underlying NASH with liver fibrosis (see Clinical Considerations ). In animal reproduction studies, adverse effects on embryo-fetal development occurred in pregnant rabbits treated with resmetirom at 3.5 times the maximum recommended dose during organogenesis. These effects were associated with maternal toxicity, whereas no embryo-fetal effects were observed at lower dose levels with better tolerance in pregnant rabbits. No embryo-fetal developmental effects occurred in pregnant rats treated with resmetirom or the metabolite MGL-3623. A pre- and postnatal development study in rats with maternal dosing of resmetirom during organogenesis through lactation showed a decrease in birthweight and increased incidence of stillbirths and mortality (postnatal days 1-4) at 37 times the maximum recommended dose (see Data ) . These effects were associated with marked suppression of maternal T4, T3, and TSH levels.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, and other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Report pregnancies to Madrigal Pharmaceuticals, Inc. Adverse Event reporting line at 1-800-905-0324 or https://pregnancyregistry.madrigalpharma.com .
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
There are risks to the mother and fetus related to underlying maternal NASH with liver fibrosis, such as increased risks of gestational diabetes, hypertensive complications, preterm birth, and postpartum hemorrhage.
Data
Animal Data
No effects on embryo-fetal development were observed in pregnant rats treated orally with up to 100 mg/kg/day (21 times the maximum recommended dose based on AUC [area under the plasma concentration-time curve]) or in pregnant rabbits treated orally with up to 30 mg/kg/day (2.8 times the maximum recommended dose based on AUC) during the period of organogenesis. Oral administration of 75 mg/kg/day in pregnant rabbits (3.5 times the maximum recommended dose based on AUC) produced an increase in post-implantation loss and decreases in viable fetuses and fetal weight. These effects were likely due to maternal toxicity (i.e., marked reductions in weight gain and food consumption).
A pre- and postnatal development study was performed using oral administration of 3, 30, or 100 mg/kg/day in female rats during organogenesis through lactation. Treatment with 100 mg/kg/day (37 times the maximum recommended dose based on AUC) produced increases in number of stillborn, pup deaths during postnatal days 1-4, and pups with absence of milk in stomach. Birthweight was decreased by 10% in this dose group, with recovery to normal body weight thereafter. The effects in offspring were associated with marked reductions in maternal plasma levels of T4 (88% decrease), T3 (79% decrease), and TSH (44% decrease). No effects on postnatal development were observed at doses up to 30 mg/kg/day (7.2 times the maximum recommended dose based on AUC). This study lacked a complete evaluation of physical and neurobehavioral development in offspring; however, no effects of resmetirom were noted in tests of learning and memory.
The metabolite MGL-3623 was tested for its effects on embryo-fetal development. No effects were observed in pregnant rats treated orally with up to 100 mg/kg/day MGL-3623 (4.7 times the maximum recommended dose based on AUC for MGL-3623) during the period of organogenesis.
8.2 Lactation
Risk Summary
There is no information regarding the presence of REZDIFFRA in human or animal milk, the effects on the breast-fed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for REZDIFFRA and any potential adverse effects on the breastfed infant from REZDIFFRA or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of REZDIFFRA have not been established in pediatric patients.
Geriatric Use
In Trial 1, of the 594 patients with NASH who received at least one dose of REZDIFFRA, 149 (25%) were 65 years of age and older and 13 (2%) were 75 years of age and older [see Clinical Studies (14) ]. No overall differences in effectiveness but numerically higher incidence of adverse reactions have been observed in patients 65 years of age and older compared to younger adult patients.
Renal Impairment
The recommended dosage of REZDIFFRA in patients with mild, moderate, or severe renal impairment is the same as in patients with normal kidney function [see Clinical Pharmacology (12.3) ] .
Hepatic Impairment
Avoid use of REZDIFFRA in patients with decompensated cirrhosis (consistent with moderate to severe hepatic impairment) . Moderate or severe hepatic impairment (Child-Pugh Class B or C) increases resmetirom C max and AUC [see Clinical Pharmacology (12.3 ) ], which may increase the risk of adverse reactions.
No dosage adjustment is recommended for patients with mild hepatic impairment (Child-Pugh Class A) [see Clinical Pharmacology (12.3) ] .
The safety and effectiveness of REZDIFFRA have not been established in patients with NASH cirrhosis.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Hepatotoxicity
Hepatotoxicity has been observed with use of REZDIFFRA. One patient had normal alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin (TB) levels at baseline, who received REZDIFFRA 80 mg daily, developed substantial elevations of liver biochemistries that resolved when treatment was interrupted. After reinitiating REZDIFFRA, the patient had elevations of ALT, AST, and TB. Peak values observed were 58 x upper limit of normal (ULN) for ALT, 66 x ULN for AST, 15 x ULN for TB, with no elevation of alkaline phosphatase (ALP). Elevations in liver enzymes were accompanied by elevations in immunoglobulin G levels, suggesting drug-induced autoimmune-like hepatitis (DI-ALH). The liver tests returned to baseline following hospitalization and discontinuation of REZDIFFRA without any therapeutic intervention.
Monitor patients during treatment with REZDIFFRA for elevations in liver tests and for the development of liver-related adverse reactions. Monitor for symptoms and signs of hepatotoxicity (e.g., fatigue, nausea, vomiting, right upper quadrant pain or tenderness, jaundice, fever, rash, and/or eosinophilia [>5%]). If hepatotoxicity is suspected, discontinue REZDIFFRA and continue to monitor the patient. If laboratory values return to baseline, weigh the potential risks against the benefits of restarting REZDIFFRA. If laboratory values do not return to baseline, consider DI-ALH or autoimmune liver disease in the evaluation of elevations in liver tests.
Gallbladder-Related Adverse Reactions
In clinical trials, cholelithiasis, acute cholecystitis, and obstructive pancreatitis (gallstone) were observed more often in REZDIFFRA-treated patients than in placebo-treated patients. If cholelithiasis is suspected, gallbladder diagnostic studies and appropriate clinical follow-up are indicated. If an acute gallbladder event is suspected, interrupt REZDIFFRA treatment until the event is resolved [see Adverse Reactions (6.1) ] .
Drug Interaction with Certain Statins
An increase in exposure of atorvastatin, pravastatin, rosuvastatin and simvastatin was observed when concomitantly administered with REZDIFFRA [see Clinical Pharmacology (12.3) ], which may increase the risk of adverse reactions related to these drugs. Dosage adjustment for certain statins is recommended [see Drug Interactions (7.2) ] . Monitor for statin-related adverse reactions including but not limited to elevation of liver tests, myopathy, and rhabdomyolysis .
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1) ]
- Gallbladder-Related Adverse Reactions [see Warnings and Precautions (5.2) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of REZDIFFRA was evaluated in two randomized, double-blind, placebo-controlled trials that enrolled a total of 2019 patients.
Trial 1
Trial 1 included patients who had noncirrhotic NASH with stages F2 and F3 fibrosis at eligibility (n=888) [see Clinical Studies (14) ] .
Adverse Reactions Leading to Discontinuations
The exposure-adjusted incidence rates (EAIRs) per 100 person-years (PY) for treatment discontinuation due to any adverse reaction were higher in the REZDIFFRA dosage arms: 4 per 100 PY, 5 per 100 PY, and 8 per 100 PY in placebo, REZDIFFRA 80 mg once daily, and REZDIFFRA 100 mg once daily arms, respectively. Diarrhea and nausea were the most common causes of treatment discontinuation.
Common Adverse Reactions
Table 1 displays EAIRs per 100 PY for the common adverse reactions that occurred in at least 5% of patients with F2 or F3 fibrosis treated in either drug arm with REZDIFFRA and were greater than that reported for placebo.
a Population includes adult patients with noncirrhotic NASH with liver fibrosis (stages F2 and F3 at eligibility). b Median exposure duration was 68 weeks for placebo, 74 weeks for REZDIFFRA 80 mg once daily, and 66 weeks for REZDIFFRA 100 mg once daily. c EAIRs are per 100 person-years (PY) where total PYs were 435, 435, and 407 for placebo, 80 mg once daily, and 100 mg once daily arms, respectively. d The EAIR per 100 PY can be interpreted as an estimated number of first occurrences of the adverse reaction of interest if 100 patients are treated for one year. Abbreviations: EAIR, exposure-adjusted incidence rate; PY, person-years; NASH, nonalcoholic steatohepatitis | |||
Adverse Reaction | Placebo N=294 n (EAIR d ) | REZDIFFRA 80 mg Once Daily N=298 n (EAIR d ) | REZDIFFRA 100 mg Once Daily N=296 n (EAIR d ) |
Diarrhea | 52 (14) | 78 (23) | 98 (33) |
Nausea | 36 (9) | 65 (18) | 51 (15) |
Pruritus | 18 (4) | 24 (6) | 36 (10) |
Vomiting | 15 (4) | 27 (7) | 30 (8) |
Constipation | 18 (4) | 20 (5) | 28 (8) |
Abdominal pain | 18 (4) | 22 (5) | 27 (7) |
Dizziness | 6 (1) | 17 (4) | 17 (4) |
Gastrointestinal Adverse Reactions
The incidence of gastrointestinal adverse reactions was higher for the REZDIFFRA drug arms compared to placebo. The EAIRs for gastrointestinal adverse reactions were 57 per 100 PY, 73 per 100 PY, and 89 per 100 PY in the placebo, REZDIFFRA 80 mg once daily, REZDIFFRA 100 mg once daily arms, respectively.
Diarrhea typically began early in treatment initiation and was mild to moderate in severity. The median time (Q1 to Q3) to a diarrheal event was 39 (2 to 195) days, 17 (3 to 70) days, and 6 (2 to 54) days in the placebo, REZDIFFRA 80 mg once daily, and REZDIFFRA 100 mg once daily arms, respectively.
Median duration of diarrhea was 9 days for placebo compared to 20 days for both REZDIFFRA 80 mg once daily and REZDIFFRA 100 mg once daily dosage arms.
Nausea also began early in treatment and was mild to moderate in severity. Among patients with nausea, the median time (Q1 to Q3) to a nausea event was 85 (24 to 347) days, 28 (2 to 162) days, and 5 (2 to 40) days in the placebo, REZDIFFRA 80 mg once daily, and REZDIFFRA 100 mg once daily arms, respectively. Median duration of nausea was 17 days, 26 days, and 28 days for patients in the placebo, REZDIFFRA 80 mg once daily, and REZDIFFRA 100 mg once daily arms, respectively.
Vomiting and abdominal pain adverse reactions were mild to moderate in severity.
Hypersensitivity Reactions
Reactions such as urticaria and rash, which may reflect drug hypersensitivity, were observed in patients receiving REZDIFFRA. The EAIRs for urticaria were 0.2 per 100 PY, 0.7 per 100 PY, and 1.5 per 100 PY in the placebo, REZDIFFRA 80 mg once daily, and REZDIFFRA 100 mg once daily arms, respectively. The EAIRs for rash were 3 per 100 PY in the placebo and REZDIFFRA 80 mg once daily arms compared to 5 per 100 PY in the REZDIFFRA 100 mg once daily arm.
Gallbladder-Related Adverse Reactions
A higher incidence of cholelithiasis, acute cholecystitis, and obstructive pancreatitis (gallstone) was observed in the treatment arms compared to placebo. However, the EAIRs for these events were less than 1 per 100 PY for all treatment arms.
Less Common Adverse Reactions
Additional adverse reactions that occurred more frequently in the REZDIFFRA arms compared to placebo, in less than 5% of patients, included decreased appetite, flatulence, abnormal feces, dysgeusia, vertigo, arrhythmia, palpitations, depression, erythema, hypoglycemia, tendinopathy, abnormal uterine bleeding.
Laboratory Abnormalities
Liver Tests
Increases in mean alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were observed in the first 4 weeks after initiating treatment with REZDIFFRA. In both REZDIFFRA dosage arms, the mean elevation in ALT and AST values was less than 1.5 times baseline at 4 weeks after treatment initiation. These values returned to baseline around 8 weeks after initiating treatment.
Table 2 presents the frequency of liver test elevations during Trial 1.
a TB elevations include patients with Gilbert syndrome. | |||
Placebo (%) | REZDIFFRA 80 mg Once Daily (%) | REZDIFFRA 100 mg Once Daily (%) | |
ALT > 3x ULN | 10 | 11 | 13 |
ALT > 5x ULN | 2 | 2 | 2 |
AST > 3x ULN | 10 | 9 | 12 |
AST > 5x ULN | 2 | 1 | 4 |
TB a > 2x ULN | 2 | 1 | 3 |
Thyroid Function Tests
A decrease in levels of prohormone free T4 (FT4) of mean 2%, 13%, and 17% was seen at 12 months in patients treated with placebo, REZDIFFRA 80 mg once daily, and REZDIFFRA 100 mg once daily, respectively, with minimal changes in active hormone T3 or in TSH. There were no clinical findings associated with FT4 decreases.
Additional Safety Data
The safety evaluation of REZDIFFRA also included an analysis of an additional randomized placebo-controlled safety trial which included 969 patients from a relevant patient population (placebo [n=318], REZDIFFRA 80 mg once daily [n=327], and REZDIFFRA 100 mg once daily [n=324]).
Data from the safety trial was combined with data from NASH patients with F2 and F3 fibrosis at eligibility (n=888) and data from an additional 162 patients from a relevant patient population enrolled in Trial 1. In the combined safety population (n=2019), the median (Q1 to Q3) age of patients at baseline was 58 (50 to 65) years; 55% were female, 28% were Hispanic, 89% were White, 2% were Asian, and 4% were Black or African American.
The safety profile from this combined analysis was similar to that in Trial 1, other than the one case of hepatotoxicity in the safety trial [see Warnings and Precautions (5.1) ] .
DRUG INTERACTIONS
Effects of Strong or Moderate CYP2C8 Inhibitors on REZDIFFRA
Table 3 includes clinically significant drug interaction effects of strong or moderate CYP2C8 inhibitors on REZDIFFRA.
| Clinical Impact | Resmetirom is a CYP2C8 substrate. Concomitant use with a strong or moderate CYP2C8 inhibitor can increase resmetirom Cmax and AUC [ see Clinical Pharmacology (12.3) ], which may increase the risk of REZDIFFRA adverse reactions. |
| Intervention | Concomitant use of REZDIFFRA with strong CYP2C8 inhibitors (e.g., gemfibrozil) is not recommended. Reduce REZDIFFRA dosage if used concomitantly with a moderate CYP2C8 inhibitor (e.g., clopidogrel) [ see Dosage and Administration (2.2) ]. |
Effects of REZDIFFRA on Other Drugs
Table 4 includes clinically significant drug interactions affecting other drugs.
Statins (Atorvastatin, Pravastatin, Rosuvastatin, or Simvastatin) | |
Clinical Impact | REZDIFFRA increased plasma concentrations of some statins (atorvastatin, pravastatin, rosuvastatin and simvastatin) [see Clinical Pharmacology (12.3) ] , which may increase the risk of adverse reactions related to these drugs. |
Intervention | Rosuvastatin and simvastatin: Limit daily statin dosage to 20 mg. Pravastatin and atorvastatin: Limit daily statin dosage to 40 mg. |
CYP2C8 Substrates | |
Clinical Impact | Resmetirom is a weak CYP2C8 inhibitor. Resmetirom increases exposure of CYP2C8 substrates [see Clinical Pharmacology (12.3) ] , which may increase the risk of adverse reactions related to these substrates. |
Intervention | Monitor patients more frequently for substrate-related adverse reactions if REZDIFFRA is co-administered with CYP2C8 substrates where minimal concentration changes may lead to serious adverse reactions. |
DESCRIPTION
REZDIFFRA (resmetirom) tablets contain resmetirom, a thyroid hormone receptor-beta agonist. The chemical name for REZDIFFRA is 2-[3,5-Dichloro-4-((6-oxo-5-(propan-2-yl)-1,6-dihydropyridazin-3-yl)oxy)phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile. The molecular formula is C 17 H 12 Cl 2 N 6 O 4 and the molecular weight is 435.22. The chemical structure is:
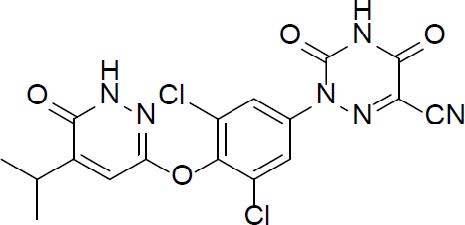
Resmetirom has low aqueous solubility below pH 6 and higher solubility above pH 7 (0.44 mg/mL at pH 7.04).
REZDIFFRA tablets are supplied in 60 mg, 80 mg, and 100 mg strengths for oral administration. Each tablet contains the active ingredient, resmetirom, and the following USP/NF excipients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, mannitol, and microcrystalline cellulose. REZDIFFRA tablets are film-coated with an Opadry coating comprised of polyethylene glycol, polyvinyl alcohol, talc, titanium dioxide, red iron oxide (100 mg tablets), yellow iron oxide (80 mg and 100 mg tablets).
CLINICAL PHARMACOLOGY
Mechanism of Action
Resmetirom is a partial agonist of the thyroid hormone receptor-beta (THR-β). Resmetirom produced 83.8% of the maximum response compared to triiodothyronine (T3), with an EC 50 of 0.21 µM in an in vitro functional assay for THR-β activation. The same functional assay for thyroid hormone receptor-alpha (THR-α) agonism showed 48.6% efficacy for resmetirom relative to T3, with an EC 50 of 3.74 µM. THR-β is the major form of THR in the liver, and stimulation of THR-β in the liver reduces intrahepatic triglycerides, whereas actions of thyroid hormone outside the liver, including in heart and bone, are largely mediated through THR-α.
Pharmacodynamics
Liver Fat Content
Resmetirom decreases liver fat content as measured by magnetic resonance imaging-protein density fat fraction (MRI-PDFF) or FibroScan controlled attenuation parameter (CAP).
Reductions in liver fat content by MRI-PDFF were observed at 16 (the first assessment) and 52 weeks of treatment. Reductions in liver fat content by CAP were observed at 52 weeks of treatment.
Prohormone FT4
Resmetirom decreased concentrations of prohormone FT4 were observed at the first assessment at 4 weeks of treatment. Similar decreases in FT4 were observed during the treatment [see Adverse Reactions (6.1) ].
Sex Hormone Binding Globulin (SHBG)
Resmetirom increased concentrations of sex hormone binding globulin (SHBG) were observed at the first assessment at 4 weeks of treatment, and at longer durations of treatment. The clinical significance of this change is unknown.
Cardiac Electrophysiology
At a dose two times the maximum recommended dose, resmetirom does not prolong the QT interval to any clinically relevant extent.
Pharmacokinetics
Following once daily doses, steady state is typically reached within 3 to 6 days of dosing. Resmetirom steady state exposure increases in a dose proportional manner between doses of 40 mg (0.5 times the lowest approved recommended dose) and 100 mg. Resmetirom exposure increases in a greater than dose proportional manner between doses of 100 mg and 200 mg (2 times the highest approved recommended dose) by about 5.6-fold. Resmetirom exposure increased 1.5- to 3-fold following once daily dosing; however, the MGL-3623 metabolite does not accumulate. The estimated resmetirom systemic exposure at steady state in NASH patients is summarized in Table 5 . Resmetirom exposure is similar between NASH patients with F2 stage fibrosis and F3 stage fibrosis.
Abbreviations: AUC t au,ss = area under the concentration-versus-time curve over one dosing interval at steady state; C max,ss = maximum concentration at steady state; CV = arithmetic coefficient of variation | ||
Parameter | Resmetirom 80 mg Once Daily Mean (CV%) | Resmetirom 100 mg Once Daily Mean (CV%) |
C max,ss (ng/mL) a | 778 (41.5) | 971 (40.9) |
AUC tau,ss (ng•h/mL) a | 5850 (60.5) | 7780 (65.5) |
Absorption
The resmetirom median time to maximum plasma concentration (T max ) is approximately 4 hours following multiple daily doses of resmetirom 80 mg or 100 mg.
Effect of Food
No clinically significant differences in resmetirom pharmacokinetics were observed following administration with a high-fat meal (approximately 150, 250, and 500-600 calories from protein, carbohydrate, and fat, respectively). Concomitant food administration resulted in a 33% decrease in C max , an 11% decrease in AUC, and a delay in median T max by about 2 hours compared to under fasted condition.
Distribution
Resmetirom apparent volume of distribution (Vd/F) at steady-state is 68 (227%) L. Resmetirom is greater than 99% protein-bound.
Elimination
Resmetirom median terminal plasma half-life (t½) is 4.5 hours and the steady state apparent clearance (CL/F) is 17.5 (56.3%) L/h.
Metabolism
Resmetirom is metabolized by CYP2C8 and is not metabolized by other CYP enzymes in vitro.
MGL-3623 is a major metabolite with a 28-times lower potency for THR-β than resmetirom. MGL-3623 represents 33% to 51% of resmetirom AUC at steady state following administration of 100 mg once daily.
Excretion
Following oral administration of a 100 mg radio-labeled dose of resmetirom, approximately 67% of the total radioactive dose was recovered in the feces, mostly as metabolites and 24% of the total radioactive dose was recovered in the urine. Unchanged labeled resmetirom was not detected in feces and accounted for 1% of the dose recovered in urine. A metabolite MGL-3623 accounted for 3.3% and 16% of the dose recovered in feces and urine, respectively. Oxalic acid metabolite was observed in plasma but not in urine.
Specific Populations
No clinically significant differences in the pharmacokinetics of resmetirom were observed based on age (18 to 83 years), sex, race (White, Black, or Asian), or ABCG2 genotype (BCRP p.Gln141Lys, p.Val12Met).
Population PK analyses indicated no clinically significant difference in the pharmacokinetics of resmetirom by mild to moderate renal impairment (eGFR 30 to 89 mL/min/1.73 m 2 , Modification of Diet in Renal Disease (MDRD)). In subjects with severe renal impairment (eGFR <30 mL/min/1.73 m², CKD-EPI), repeated 100 mg once daily dosing of resmetirom resulted in an approximately 1.5-fold increase in resmetirom AUC and a 1.3-fold increase in resmetirom C max compared to matched healthy subjects. In the same study, MGL-3623 AUC tau was 1.4-fold higher and C max was 1.06-fold higher in patients with severe renal impairment, compared to subjects with normal renal function.
Body Weight
A clinically significant difference in resmetirom exposure was not observed with the recommended weight-based dosage . However, resmetirom CL/F and Vd/F increase with increasing body weight, resulting in lower resmetirom exposure in patients with higher body weight receiving the same dosage as lower weight patients [see Dosage and Administration (2.1) ] .
Patients with Hepatic Impairment
Patients with Hepatic Impairment: Following repeated 80 mg once daily dosing of resmetirom for 6 days, resmetirom AUC was 1.3-fold, 2.7-fold and 19-fold higher in patients with mild, moderate and severe hepatic impairment (Child-Pugh A, B and C), respectively compared to subjects with normal hepatic function. Resmetirom C max was 1.2-fold, 1.7-fold and 8.1-fold higher in patients with mild, moderate and severe hepatic impairment, respectively, compared to subjects with normal hepatic function (Table 6 ).
In the same study, MGL-3623 AUC tau was 1.3-fold, 2-fold and 5.8-fold higher in patients with mild, moderate and severe hepatic impairment, respectively, compared to subjects with normal hepatic function [see Use in Specific Populations (8.7) ].
a Exposure parameters presented as Mean (CV%) b N = 8 Abbreviations: AUC tau,ss = area under the concentration-versus-time curve over one dosing interval at steady state; C max,ss = maximum concentration at steady state; CV = arithmetic coefficient of variation | ||||
Parameter | Normal Hepatic Function (N = 7) | Child-Pugh Class | ||
A Mild (N = 10) | B Moderate (N = 9) | C Severe (N = 3) | ||
Resmetirom | ||||
| C max,ss (ng/mL) a | 1070 (51.0) | 1390 (67.8) | 1830 (47.5) | 7730 (17.4) |
| AUC tau,ss (ng•h/mL) a | 5100 (51.5) | 5570 (66.4) | 15100 (65.8) b | 97600 (39.0) |
NASH Patients with Mild Hepatic Impairment (Child-Pugh A): Geometric mean AUC and C max in NASH cirrhosis patients with mild hepatic impairment (Child-Pugh Class A; n = 20) were 6% higher and 10% lower, respectively, compared to non-cirrhotic NASH patients following repeated 100 mg once daily dosing of resmetirom for 6 days. The safety and effectiveness of resmetirom have not been established in patients with NASH cirrhosis.
Drug Interaction Studies
Clinical Studies
Moderate CYP2C8 Inhibitors: Resmetirom C max increased 1.3-fold and AUC 1.7-fold following concomitant use of multiple doses of resmetirom 100 mg/day with clopidogrel (a moderate CYP2C8 inhibitor) at steady-state in healthy subjects [see Drug Interactions (7.1) ] .
CYP2C8 Substrates: Pioglitazone (a CYP2C8 substrate) C max was unchanged and AUC increased 1.5-fold following concomitant use of a single oral dose of pioglitazone (15 mg) with resmetirom at steady state (100 mg/day) in healthy subjects [see Drug Interactions (7.1) ] .
Simvastatin: Simvastatin (OATP1B1 and OATP1B3 substrate) C max increased 1.4-fold and AUC 1.7- fold following concomitant use of a single oral dose of simvastatin (20 mg) with resmetirom at steady state (100 mg/day) in healthy subjects [see Drug Interactions (7.2) ] .
Rosuvastatin: Rosuvastatin (BCRP, OATP1B1, and OATP1B3 substrate) C max increased 1.8-fold and AUC 1.4-fold following concomitant use of a single oral dose of rosuvastatin (20 mg) with resmetirom at steady state (100 mg/day) in healthy subjects [see Drug Interactions (7.2) ] .
Pravastatin: Pravastatin (OATP1B1 and OATP1B3 substrate) C max increased 1.3-fold and AUC 1.4-fold following concomitant use of a single oral dose of pravastatin (40 mg) with resmetirom at steady state (100 mg/day) in healthy subjects [see Drug Interactions (7.2) ] .
Atorvastatin: Atorvastatin (BCRP, OATP1B1, and OATP1B3 substrate) C max was unchanged and AUC increased 1.4-fold following concomitant use of a single oral dose of atorvastatin (20 mg) with resmetirom at steady state (100 mg/day) in healthy subjects. Atorvastatin lactone C max increased 2.0-fold and AUC increased 1.8-fold [see Drug Interactions (7.2) ] .
OATP1B1, OATP1B3 and BCRP inhibitors: No clinically significant differences in the pharmacokinetics of resmetirom were observed when used concomitantly with cyclosporine (an OATP1B1/1B3 and BCRP inhibitor).
Other Drugs: No clinically significant differences in the pharmacokinetics of R-warfarin or S-warfarin were observed when used concomitantly with resmetirom.
In Vitro Studies
CYP450 Enzymes: Resmetirom is an inhibitor of CYP2C8.
Glucuronidation Enzymes: Resmetirom is an inhibitor of UDP-glucuronosyltransferases (UGTs) 1A4 and 1A9. The clinical relevance of UGT1A4 and UGT1A9 inhibition is unknown.
Transporters: Resmetirom is a substrate for organic anion-transporting polypeptides (OATP) 1B1 and 1B3 and breast cancer resistance protein (BCRP). Resmetirom inhibits OATP1B1, OATP1B3, BCRP, OAT3, and bile salt export pump (BSEP). The clinical significance of OAT3 and BSEP inhibition is unknown.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 2-year study in CD-1 mice, resmetirom produced leiomyoma or leiomyosarcoma in the uterus at a dose of 100 mg/kg/day (51 times the maximum recommended dose based on AUC). No tumorigenic effects were observed in female mice at doses of up to 30 mg/kg/day (14 times the maximum recommended dose based on AUC) or in male mice at doses of up to 100 mg/kg/day (35 times the maximum recommended dose based on AUC).
In a 2-year study in Sprague-Dawley rats, resmetirom produced benign fibroadenoma in the mammary gland of males at a dose of 30 mg/kg/day (6.5 times the maximum recommended dose based on AUC). No tumorigenic effects were observed in male rats at doses of up to 6 mg/kg/day (3.7 times the maximum recommended dose based on AUC) or in female rats at doses of up to 30 mg/kg/day (3.4 times the maximum recommended dose based on AUC).
In a 26-week study in transgenic [CByB6F1-Tg(HRAS)2Jic] mice, the major metabolite of resmetirom, MGL-3623, was not tumorigenic at doses of up to 1500 mg/kg/day.
Mutagenesis
Resmetirom was negative in the in vitro bacterial reverse mutation (Ames) assay, the in vitro chromosomal aberration assay in human peripheral blood lymphocytes, the in vitro micronucleus assay in L5178Y tk +/- mouse lymphoma cells, and the in vivo rat micronucleus assay.
The metabolite MGL-3623 was negative in the in vitro bacterial reverse mutation (Ames) assay and the in vivo rat micronucleus assay. MGL-3623 tested positive in the presence of metabolic activation in the in vitro micronucleus assay with TK6 human lymphoblast cells, with the increase in micronuclei limited to a single concentration that produced 59% growth inhibition.
Impairment of Fertility
Resmetirom had no effects on fertility or reproductive function in male and female rats at oral doses of up to 30 mg/kg/day (6.9 times and 2.6 times the maximum recommended dose in male and female rats, respectively, based on AUC).
CLINICAL STUDIES
The efficacy of REZDIFFRA was evaluated based on an efficacy analysis at Month 12 in Trial 1 (NCT03900429 ), a 54-month, randomized, double-blind, placebo-controlled trial. Enrolled patients had metabolic risk factors and a baseline or recent liver biopsy showing NASH with fibrosis stage 2 or 3 and a NAFLD Activity Score (NAS) of at least 4. Efficacy determination was based on the effect of REZDIFFRA on resolution of steatohepatitis without worsening of fibrosis and one stage improvement in fibrosis without worsening of steatohepatitis, on post-baseline liver biopsies collected at 12 months.
The month 12 analysis included 888 F2 and F3 (at eligibility) patients randomized 1:1:1 to receive placebo (n = 294), REZDIFFRA 80 mg once daily (n = 298), or REZDIFFRA 100 mg once daily (n = 296), in addition to lifestyle counseling on nutrition and exercise. Patients were on stable doses of medications for diabetes, dyslipidemia, and hypertension.
Demographic and baseline characteristics were balanced between treatment and placebo groups. Overall, the median (Q1 to Q3) age of patients at baseline was 58 (51 to 65) years, 56% were female, 21% were Hispanic, 89% were White, 3% were Asian, and 2% were Black or African American. Median (Q1 to Q3) body mass index (BMI) was 35 (31 to 40) kg/m 2 and median (Q1 to Q3) body weight was 99 (85 to 114) kg. Baseline characteristics are presented in Table 7 .
a Less than 5% missingness in these variables is omitted. b kPa = kilopascal; Db/M = decibels per meter | |
Characteristic | Overall N=888 |
Fibrosis stage, n (%) F2 F3 | |
328 (37) 560 (63) | |
Type 2 Diabetes, n (%) | 608 (68) |
Hypertension, n (%) | 700 (79) |
Dyslipidemia, n (%) | 633 (71) |
Statin use, n (%) | 434 (49) |
Thyroxine use, n (%) | 124 (14) |
Vibration-controlled Transient Elastography (VCTE) (kPa), Median (Q1, Q3) a, b | 12 (10, 15) |
Controlled attenuation parameter (CAP) (Db/M), Median (Q1, Q3) a | 349 (320, 378) |
Fibrosis Index Based on 4 Factors (FIB-4), Median (Q1, Q3) a | 1.3 (1, 1.8) |
Enhanced Liver Fibrosis (ELF), Median (Q1, Q3) a | 9.7 (9.2, 10.4) |
Table 8 presents the Month 12 histopathology results comparing REZDIFFRA with placebo on 1) the percentage of patients with resolution of steatohepatitis and no worsening of liver fibrosis and 2) the percentage of patients with at least one stage improvement in liver fibrosis and no worsening of steatohepatitis. Two pathologists, Pathologist A and Pathologist B, independently read the liver biopsies for each patient. Both the 80 mg once daily and the 100 mg once daily dosages of REZDIFFRA demonstrated improvement on these histopathology endpoints at Month 12 compared to placebo. In a statistical analysis incorporating both pathologists’ independent readings, REZDIFFRA achieved statistical significance on both histopathology endpoints for both doses.
Examination of age, gender, diabetes status (Yes or No), and fibrosis stage (F2 or F3) subgroups did not identify differences in response to REZDIFFRA among these subgroups. The majority of patients in the trial were white (89%); there were too few patients of other races to adequately assess differences in response by race.
Liver fibrosis was evaluated on the NASH Clinical Research Network (CRN) fibrosis score as 0 to 4. Resolution of steatohepatitis was defined as a score of 0–1 for inflammation, 0 for ballooning, and any value for steatosis. No worsening of steatohepatitis was defined as no increase in score for ballooning, inflammation, or steatosis. Estimated using the Mantel-Haenszel method stratified by baseline type 2 diabetes status (presence or absence) and fibrosis stage (F2 or F3). 95% stratified Newcombe confidence intervals (CIs) are provided. Patients with missing liver biopsy at Month 12 are considered a non-responder. | |||
Placebo N=294 | REZDIFFRA 80 mg Once Daily N=298 | REZDIFFRA 100 mg Once Daily N=296 | |
Resolution of steatohepatitis and no worsening of liver fibrosis | |||
Response rate, Pathologist A (%) | 13 | 27 | 36 |
Difference in response rate vs. placebo (95% CI) | 14 (8, 20) | 23 (16, 30) | |
Response rate, Pathologist B (%) | 9 | 26 | 24 |
Difference in response rate vs. placebo (95% CI) | 17 (11, 23) | 15 (9, 21) | |
Improvement in liver fibrosis and no worsening of steatohepatitis | |||
Response rate, Pathologist A (%) | 15 | 23 | 28 |
Difference in response rate vs. placebo (95% CI) | 8 (2, 14) | 13 (7, 20) | |
Response rate, Pathologist B (%) | 13 | 23 | 24 |
Difference in response rate vs. placebo (95% CI) | 11 (5, 17) | 11 (5, 17) | |
Starting at Month 3 and through Month 12, there was a trend of greater reductions from baseline in average ALT and AST in the REZDIFFRA groups as compared to the placebo group.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
REZDIFFRA (resmetirom) tablets are packaged in white high-density polyethylene bottles closed with a child-resistant closure containing an induction seal.
60 mg Tablets : white oval-shaped film-coated tablets, debossed “P60” on one side and plain on the other side.
- Bottle of 30 count (NDC 82576-060-30)
80 mg Tablets : yellow, oval-shaped, film-coated tablets, debossed with “P80” on one side and plain on the other side.
- Bottle of 30 count (NDC 82576-080-30)
- Bottle of 90 count (NDC 82576-080-90)
100 mg Tablets: beige to pink, oval-shaped, film-coated tablets, debossed with “P100” on one side and plain on the other side.
- Bottle of 30 count (NDC 82576-100-30)
- Bottle of 90 count (NDC 82576-100-90)
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [ see USP Controlled Room Temperature ].