Get your patient on Simponi (Golimumab)
Simponi patient education
Patient toolkit
Dosage & administration

Simponi prescribing information
WARNING: SERIOUS INFECTIONS AND MALIGNANCY
SERIOUS INFECTIONS
Patients treated with SIMPONI are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1) ] . Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
Discontinue SIMPONI if a patient develops a serious infection.
Reported infections with TNF blockers, of which SIMPONI is a member, include:
- Active tuberculosis, including reactivation of latent tuberculosis. Patients with tuberculosis have frequently presented with disseminated or extrapulmonary disease. Test patients for latent tuberculosis before SIMPONI use and during therapy. Initiate treatment for latent TB prior to SIMPONI use.
- Invasive fungal infections including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Consider empiric antifungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella and Listeria.
Consider the risks and benefits of treatment with SIMPONI prior to initiating therapy in patients with chronic or recurrent infection.
Monitor patients closely for the development of signs and symptoms of infection during and after treatment with SIMPONI, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1) ] .
MALIGNANCY
Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, of which SIMPONI is a member [see Warnings and Precautions (5.2) ] .
INDICATIONS AND USAGE
SIMPONI is a tumor necrosis factor (TNF) blocker indicated for the treatment of:
- adult patients with moderately to severely active rheumatoid arthritis (RA) in combination with methotrexate (1.1 )
- adult patients with active psoriatic arthritis (PsA) alone, or in combination with methotrexate (1.2 )
- adult patients with active ankylosing spondylitis (AS) (1.3 )
- adult and pediatric patients weighing at least 15 kg with moderately to severely active ulcerative colitis (UC) (1.4 )
Rheumatoid Arthritis
SIMPONI, in combination with methotrexate, is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis.
Psoriatic Arthritis
SIMPONI, alone or in combination with methotrexate, is indicated for the treatment of adult patients with active psoriatic arthritis.
Ankylosing Spondylitis
SIMPONI is indicated for the treatment of adult patients with active ankylosing spondylitis.
Ulcerative Colitis
SIMPONI is indicated for the treatment of adults and pediatric patients weighing at least 15 kg with moderately to severely active ulcerative colitis .
DOSAGE AND ADMINISTRATION
- RA, PsA, and AS : 50 mg administered by subcutaneous injection once a month (2.2 )
- UC : The recommended dosage and administration by subcutaneous injection in adults and pediatric patients weighing at least 15 kg is shown in the table (2.3 )
| Recommended Dosage | |||
|---|---|---|---|
| Weight for Patients with UC | Week 0 | Week 2 | Week 6 and every 4 weeks thereafter |
| Adults and pediatric patients 40 kg and greater For pediatric patients weighing 15 kg or greater, administer the appropriate dose using the prefilled syringe (50 mg/0.5 mL or 100 mg/mL). | 200 mg | 100 mg | 100 mg |
| Pediatric patients at least 15 kg to less than 40 kg | 100 mg | 50 mg | 50 mg |
Recommended Evaluations and Immunizations Before Initiating SIMPONI
Prior to initiating treatment with SIMPONI:
- Evaluate patients for active tuberculosis and test for latent infection [see Warnings and Precautions (5.1) ].
- Test patients for hepatitis B viral infection.
- If possible, complete all age-appropriate vaccinations according to current immunization guidelines [see Warnings and Precautions (5.11) ] .
Recommended Dosage for Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis
The recommended SIMPONI dosage in adults is 50 mg administered by subcutaneous injection once a month.
For patients with rheumatoid arthritis (RA), SIMPONI should be given in combination with methotrexate and for patients with psoriatic arthritis (PsA) or ankylosing spondylitis (AS), SIMPONI may be given with or without methotrexate or other nonbiologic Disease-Modifying Antirheumatic Drugs (DMARDs). For patients with RA, PsA, or AS, corticosteroids, non-biologic DMARDs, and/or NSAIDs may be continued during treatment with SIMPONI.
Recommended Dosage for Moderately to Severely Active Ulcerative Colitis in Adults and Pediatric Patients Weighing at least 15 kg
The recommended dosage is shown in Table 1.
| Weight for Patients with UC | Recommended Dosage of SIMPONI | ||
|---|---|---|---|
| Week 0 | Week 2 | Week 6 and every 4 weeks thereafter | |
| Adults and pediatric patients 40 kg and greater For pediatric patients weighing 15 kg or greater, administer the appropriate dose using the prefilled syringe (50 mg/0.5 mL or 100 mg/mL). | 200 mg | 100 mg | 100 mg |
| Pediatric patients at least 15 kg to less than 40 kg | 100 mg | 50 mg | 50 mg |
Preparation and Administration Instructions
SIMPONI is intended for use under the guidance and supervision of a healthcare provider after proper training in subcutaneous injection technique. Patients may self-inject with SIMPONI if a physician determines that it is appropriate. Instruct patients to follow the directions provided in the Instructions for Use.
SIMPONI Prefilled Syringe
- Adult and pediatric patients 12 years of age and older may self-inject with SIMPONI prefilled syringe.
SIMPONI SmartJect ® Autoinjector
- Adult patients may self-inject with SIMPONI SmartJect ® autoinjector.
- Use of the SmartJect ® autoinjector for pediatric self-administration has not been evaluated.
• To ensure proper use, allow the prefilled syringe or autoinjector to sit at room temperature outside the carton for at least 30 minutes prior to subcutaneous injection. Do not warm SIMPONI in any other way.
• Prior to administration, visually inspect the solution for particles and discoloration through the viewing window. SIMPONI is clear to slightly opalescent and colorless to light yellow. Do not use SIMPONI, if the solution is discolored, or cloudy, or if foreign particles are present.
• Do not use any leftover product remaining in the prefilled syringe or prefilled autoinjector.
• Instruct patients sensitive to latex not to handle the needle cover on the prefilled syringe or the needle cover of the prefilled syringe within the autoinjector cap because it contains dry natural rubber (a derivative of latex).
• At the time of dosing, if multiple injections are required, administer the injections at different sites on the body.
• Rotate injection sites and never give injections into areas where the skin is tender, bruised, red, or hard.
• If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
DOSAGE FORMS AND STRENGTHS
Injection: 50 mg/0.5 mL and 100 mg/mL clear to slightly opalescent, colorless to light yellow solution in a single-dose prefilled syringe or single-dose SmartJect ® autoinjector.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Available data from postmarketing case reports with golimumab use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. An observational study of northern European births observed similar unadjusted rates of major birth defects in infants exposed in utero to golimumab compared to no treatment or non-biologic systemic therapy. However, this study had important limitations (see Data ) .
Monoclonal antibodies, such as golimumab, are transported across the placenta during the third trimester of pregnancy and may affect immune response in the in utero exposed infant (see Clinical Considerations ) . In an animal reproductive study, golimumab administered by the subcutaneous route to pregnant monkeys, during the period of organogenesis, at doses that produced exposures approximately 360 times the maximum recommended human dose (MRHD) had no adverse fetal effects (see Data ) . In a pre- and post-natal development study with pregnant monkeys, subcutaneous administration of golimumab, during the later gestational and lactation periods, at doses producing maximal maternal blood concentrations approximately 460 times those found with the MRHD had no adverse developmental effects on infants (see Data ) . Data suggest that there are risks to the mother and the fetus associated with rheumatoid arthritis and ulcerative colitis in pregnancy (see Clinical Considerations ) .
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and of miscarriage is 15–20%, respectively.
Clinical Considerations
Disease-associated Maternal and/or Embryo/Fetal Risk
Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or ulcerative colitis. Adverse pregnancy outcomes include preterm delivery (before 37 weeks gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Fetal/Neonatal Adverse Reactions
Golimumab crosses the placenta during pregnancy. Another TNF-blocking monoclonal antibody administered during pregnancy was detected for up to 6 months in the serum of infants. Consequently, these infants may be at increased risk of infection. Administration of live vaccines to infants exposed to SIMPONI in utero is not recommended for 6 months following the mother's last SIMPONI injection during pregnancy [see Warnings and Precautions (5.11) and Drug Interactions (7.3) ] .
Data
Human Data
An observational, exposure-based, cohort study based on data from the Swedish, Danish, and Finnish Medical Birth Registers conducted between 2006–2020 (Sweden and Denmark) and 2006–2019 (Finland) compared the risk of major birth defects in 134 live-born infants exposed to golimumab (116 from women treated for rheumatic conditions, 18 from women treated for ulcerative colitis) to no treatment or non-biologic systemic therapy. The unadjusted rate of major birth defects in infants exposed in utero was similar across all groups. However, this study had important limitations such as a small number of pregnant women exposed to golimumab, a wide exposure ascertainment window, and incomplete risk adjustment for potential confounders.
Animal Data
In an embryofetal developmental toxicology study in which pregnant cynomolgus monkeys were treated with golimumab during the period organogenesis from gestation days (GD) 20 to 51, exposures up to 360 times greater than the exposure at the MRHD (on an area under the curve (AUC) basis with maternal subcutaneous doses up to 50 mg/kg twice weekly) produced no evidence of fetal malformations or embryotoxicity. There was no evidence of maternal toxicity. Umbilical cord blood samples collected at the end of the second trimester showed that fetuses were exposed to golimumab during gestation.
In a pre- and post-natal developmental study in which pregnant cynomolgus monkeys were treated with golimumab from gestation day 50 to postpartum day 33, maximal drug concentrations approximately 460 times greater than that found with the MRHD (on a maximum blood concentration (C max ) basis at steady state with maternal subcutaneous doses up to 50 mg/kg twice weekly) were not associated with any evidence of developmental defects in infants. There was no evidence of maternal toxicity. Golimumab was present in fetal serum at the end of the second trimester and in neonatal serum from the time of birth and for up to 6 months postpartum.
Lactation
Risk Summary
There is no information regarding the presence of SIMPONI in human milk, the effects on breastfed infants, or the effects on milk production. Maternal IgG is known to be present in human milk. Golimumab is present in the milk of lactating cynomolgus monkeys (see Data ) . If golimumab is transferred into human milk, the effects of local exposure in the gastrointestinal tract and potential limited systemic exposure in the infant to golimumab are unknown. The developmental and health benefits of breast-feeding should be considered along with the mother's clinical need for SIMPONI and any potential adverse effects on the breast-fed infants from SIMPONI, or from the underlying maternal condition.
Data
In the pre- and post-natal development study in cynomolgus monkeys in which golimumab was administered subcutaneously during pregnancy and lactation, golimumab was detected in the breast milk at concentrations that were approximately 400-fold lower than the maternal serum concentrations.
Pediatric Use
Ulcerative Colitis
The safety and effectiveness of SIMPONI for the treatment of moderately to severely active ulcerative colitis have been established in pediatric patients weighing at least 15 kg. Use of SIMPONI for this indication is supported by evidence from adequate and well-controlled studies in adults with additional safety and efficacy data from an open-label, study in 69 pediatric patients (4 to 17 years of age). The adverse reaction profile in these pediatric patients was similar to adults [see Adverse Reactions (6.1) , Clinical Pharmacology (12.3) , and Clinical Studies (14.5) ] . Additionally, headache (17%) and pyrexia (10%) were reported in at least 10% of pediatric patients in the trial.
The safety and effectiveness of SIMPONI for the treatment of moderately to severely active ulcerative colitis have not been established in pediatric patients weighing less than 15 kg.
Polyarticular Juvenile Idiopathic Arthritis (pJIA)
The safety and efficacy of SIMPONI were evaluated in a multicenter, placebo-controlled, double-blind, randomized-withdrawal, parallel group study in 173 children (2 to 17 years of age) with active polyarticular juvenile idiopathic arthritis (pJIA) despite treatment with MTX for at least 3 months. Subjects were maintained on their stable dose of MTX at the same dose (mg/week) at study entry. Concurrent use of stable doses of oral corticosteroids (≤10 mg/day or 0.2 mg/kg/day prednisone or equivalent, whichever was less) and/or NSAIDs was permitted. In the 16 week open-label phase, all patients received MTX and SIMPONI 30 mg/m 2 (maximum 50 mg) subcutaneously every 4 weeks. Patients who achieved an ACR Ped 30 response at Week 16 entered the randomized-withdrawal phase of the study and received MTX and either SIMPONI 30 mg/m 2 (maximum 50 mg) or placebo every 4 weeks through Week 48.
The primary endpoint of the study was the proportion of patients who did not experience a flare between Week 16 and Week 48, among all subjects who entered the randomized withdrawal phase. The efficacy of SIMPONI in the treatment of pJIA was not demonstrated in this study because there was no statistical evidence of differences in flare rate between SIMPONI-treated patients and placebo patients between Weeks 16 and 48.
In this study, the frequency and type of the adverse reactions seen in children were generally similar to those observed in adults.
Geriatric Use
In the Phase 3 trials in RA, PsA, and AS, there were no overall differences in SAEs, serious infections, and AEs in SIMPONI-treated patients ages 65 or older (N=155) compared with younger SIMPONI-treated patients. Clinical studies of SIMPONI in patients with moderately to severely active ulcerative colitis did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently than younger adult patients. Because there is a higher incidence of infections in the geriatric population in general, caution should be used in treating geriatric patients with SIMPONI [see Warnings and Precautions (5.1 , 5.5) ] .
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Serious Infections: Do not start SIMPONI during an active infection. If an infection develops, monitor carefully, and stop SIMPONI if infection becomes serious (5.1 )
- Invasive Fungal Infections: For patients who develop a systemic illness on SIMPONI, consider empiric antifungal therapy for those who reside in or travel to regions where mycoses are endemic (5.1 )
- Malignancies: Incidence of lymphoma was greater than in the general U.S. population. Cases of other malignancies have been observed among patients receiving TNF blockers (5.2 )
- Congestive Heart Failure: Worsening, or new onset, may occur. Stop SIMPONI if new or worsening symptoms occur (5.3 )
- Demyelinating Disorders: Exacerbation or new onset may occur (5.4 )
- Hepatitis B Reactivation: Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop SIMPONI and begin antiviral therapy (5.5 )
- Lupus-like Syndrome: Discontinue SIMPONI if symptoms develop (5.6 )
- Hypersensitivity Reactions: Serious systemic hypersensitivity reactions including anaphylaxis may occur (5.12 )
Serious Infections
Patients treated with SIMPONI are at increased risk for developing serious infections involving various organ systems and sites that may lead to hospitalization or death.
Opportunistic infections due to bacterial, mycobacterial, invasive fungal, viral, or parasitic organisms including aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, histoplasmosis, legionellosis, listeriosis, pneumocystosis, and tuberculosis have been reported with TNF blockers. Patients have frequently presented with disseminated rather than localized disease. The concomitant use of a TNF blocker and abatacept or anakinra was associated with a higher risk of serious infections; therefore, the concomitant use of SIMPONI and these biologic products is not recommended [see Warnings and Precautions (5.6 , 5.7) and Drug Interactions (7.2) ] .
Treatment with SIMPONI should not be initiated in patients with an active infection, including clinically important localized infections. Patients greater than 65 years of age, patients with co-morbid conditions and/or patients taking concomitant immunosuppressants such as corticosteroids or methotrexate may be at greater risk of infection. Consider the risks and benefits of treatment prior to initiating SIMPONI in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses, such as histoplasmosis, coccidioidomycosis, or blastomycosis; or
- with underlying conditions that may predispose them to infection.
Monitoring
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with SIMPONI. Discontinue SIMPONI if a patient develops a serious infection, an opportunistic infection, or sepsis. For a patient who develops a new infection during treatment with SIMPONI, perform a prompt and complete diagnostic workup appropriate for an immunocompromised patient, initiate appropriate antimicrobial therapy, and closely monitor them.
Serious Infection in Clinical Trials
In controlled Phase 3 trials through Week 16 in patients with RA, PsA, and AS, serious infections were observed in 1.4% of SIMPONI-treated patients and 1.3% of control-treated patients. In the controlled Phase 3 trials through Week 16 in patients with RA, PsA, and AS, the incidence of serious infections per 100 patient-years of follow-up was 5.7 (95% CI: 3.8, 8.2) for the SIMPONI group and 4.2 (95% CI: 1.8, 8.2) for the placebo group. In the controlled Phase 2/3 trial through Week 6 of SIMPONI induction in UC, the incidence of serious infections in SIMPONI 200/100 mg-treated patients was similar to the incidence of serious infections in placebo-treated patients. Through Week 60, the incidence of serious infections was similar in patients who received SIMPONI induction and 100 mg during maintenance compared with patients who received SIMPONI induction and placebo during the maintenance portion of the UC trial. Serious infections observed in SIMPONI-treated patients included sepsis, pneumonia, cellulitis, abscess, tuberculosis, invasive fungal infections, and hepatitis B infection.
Tuberculosis
Cases of reactivation of tuberculosis or new tuberculosis infections have been observed in patients receiving TNF blockers, including patients who have previously received treatment for latent or active tuberculosis. Evaluate patients for tuberculosis risk factors and test for latent infection prior to initiating SIMPONI and periodically during therapy.
Treatment of latent tuberculosis infection prior to therapy with TNF blockers has been shown to reduce the risk of tuberculosis reactivation during therapy. Prior to initiating SIMPONI, assess if treatment for latent tuberculosis is needed; an induration of 5 mm or greater is a positive tuberculin skin test, even for patients previously vaccinated with Bacille Calmette-Guerin (BCG).
Consider anti-tuberculosis therapy prior to initiation of SIMPONI in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but having risk factors for tuberculosis infection. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Cases of active tuberculosis have occurred in patients treated with SIMPONI during and after treatment for latent tuberculosis. Monitor patients for the development of signs and symptoms of tuberculosis including patients who tested negative for latent tuberculosis infection prior to initiating therapy, patients who are on treatment for latent tuberculosis, or patients who were previously treated for tuberculosis infection.
Consider tuberculosis in the differential diagnosis in patients who develop a new infection during SIMPONI treatment, especially in patients who have previously or recently traveled to countries with a high prevalence of tuberculosis, or who have had close contact with a person with active tuberculosis.
In the controlled and uncontrolled portions of the Phase 2 RA and Phase 3 RA, PsA, and AS trials, the incidence of active TB was 0.23 and 0 per 100 patient-years in 2347 SIMPONI-treated patients and 674 placebo-treated patients, respectively. Cases of TB included pulmonary and extrapulmonary TB. The overwhelming majority of the TB cases occurred in countries with a high incidence rate of TB. In the controlled Phase 2/3 trial of SIMPONI induction through Week 6 in UC, no cases of TB were observed in SIMPONI 200/100 mg-treated patients or in placebo-treated patients. Through Week 60, the incidence per 100 patient-years of TB in patients who received SIMPONI induction and 100 mg during the maintenance portion of the UC trial was 0.52 (95% CI: 0.11, 1.53). One case of TB was observed in the placebo maintenance group in a patient who received SIMPONI intravenous (IV) induction.
Invasive Fungal Infections
If patients develop a serious systemic illness and they reside or travel in regions where mycoses are endemic, consider invasive fungal infection in the differential diagnosis. Consider appropriate empiric antifungal therapy, and take into account both the risk for severe fungal infection and the risks of antifungal therapy while a diagnostic workup is being performed. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. To aid in the management of such patients, consider consultation with a physician with expertise in the diagnosis and treatment of invasive fungal infections.
Malignancies
Malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with TNF-blocking agents (initiation of therapy ≤ 18 years of age), of which SIMPONI is a member. Approximately half the cases were lymphomas, including Hodgkin's and non-Hodgkin's lymphoma. The other cases represented a variety of malignancies, including rare malignancies that are usually associated with immunosuppression, and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months (range 1 to 84 months) after the first dose of TNF-blocker therapy. Most of the patients were receiving concomitant immunosuppressants. These cases were reported postmarketing and are derived from a variety of sources, including registries and spontaneous postmarketing reports.
The risks and benefits of TNF-blocker treatment, including SIMPONI, should be considered prior to initiating therapy in patients with a known malignancy other than a successfully treated nonmelanoma skin cancer (NMSC) or when considering continuing a TNF-blocker in patients who develop a malignancy.
In the controlled portions of clinical trials of TNF blockers, including SIMPONI, more cases of lymphoma have been observed among patients receiving anti-TNF treatment compared with patients in the control groups. During the controlled portions of the Phase 2 trials in RA, and the Phase 3 trials in RA, PsA and AS, the incidence of lymphoma per 100 patient-years of follow-up was 0.21 (95% CI: 0.03, 0.77) in the combined SIMPONI group compared with an incidence of 0 (95% CI: 0, 0.96) in the placebo group. In the controlled and uncontrolled portions of these clinical trials in 2347 SIMPONI-treated patients with a median follow-up of 1.4 years, the incidence of lymphoma was 3.8-fold higher than expected in the general U.S. population according to the 1964–2004 data from SEER database (adjusted for age, gender, and race). Through Week 60 of the UC trials, there were no cases of lymphoma with SIMPONI. Patients with RA and other chronic inflammatory diseases, particularly patients with highly active disease and/or chronic exposure to immunosuppressant therapies, may be at higher risk (up to several fold) than the general population for the development of lymphoma, even in the absence of TNF-blocking therapy. Cases of acute and chronic leukemia have been reported with TNF-blocker use, including SIMPONI, in rheumatoid arthritis and other indications. Even in the absence of TNF-blocker therapy, patients with rheumatoid arthritis may be at a higher risk (approximately 2-fold) than the general population for the development of leukemia.
Rare postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL) have been reported in patients treated with TNF-blocking agents. This rare type of T-cell lymphoma has a very aggressive disease course and is usually fatal. Nearly all of the reported TNF blocker associated cases have occurred in patients with Crohn's disease or ulcerative colitis. The majority were in adolescent and young adult males. Almost all these patients had received treatment with azathioprine (AZA) or 6-mercaptopurine (6–MP) concomitantly with a TNF blocker at or prior to diagnosis. The potential risk with the combination of AZA or 6-MP and SIMPONI should be carefully considered. A risk for the development for hepatosplenic T-cell lymphoma in patients treated with TNF blockers cannot be excluded.
During the controlled portions of the Phase 2 trial in RA, and the Phase 3 trials in RA, PsA and AS, the incidence of malignancies other than lymphoma per 100 patient-years of follow-up was not elevated in the combined SIMPONI group compared with the placebo group. In the controlled and uncontrolled portions of these trials, the incidence of malignancies, other than lymphoma, in SIMPONI-treated patients was similar to that expected in the general U.S. population according to the 1969–2004 SEER database (adjusted for age, gender, and race). In the 6-week placebo-controlled portions of the SIMPONI Phase 2/3 clinical trials in UC, the incidence of non-lymphoma malignancies (excluding nonmelanoma skin cancer) was similar between the SIMPONI and the placebo group. Through Week 60, the incidence of non-lymphoma malignancies (excluding nonmelanoma skin cancer) was similar to the general U.S. population according to the 1969–2004 SEER database (adjusted for age, gender, and race). Short follow-up periods, such as those of one year or less in the studies above, may not adequately reflect the true incidence of malignancies .
It is not known if SIMPONI treatment influences the risk for developing dysplasia or colon cancer. All patients with ulcerative colitis who are at increased risk for dysplasia or colon carcinoma (for example, patients with long-standing ulcerative colitis or primary sclerosing cholangitis), or who had a prior history of dysplasia or colon carcinoma should be screened for dysplasia at regular intervals before therapy and throughout their disease course. This evaluation should include colonoscopy and biopsies per local recommendations. In patients with newly diagnosed dysplasia treated with SIMPONI, the risks and benefits to the individual patient must be carefully reviewed and consideration should be given to whether therapy should be continued.
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF-blocking agents, including SIMPONI. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
In controlled trials of other TNF blockers in patients at higher risk for malignancies (e.g., patients with chronic obstructive pulmonary disease [COPD], patients with Wegener's granulomatosis treated with concomitant cyclophosphamide) a greater portion of malignancies occurred in the TNF-blocker group compared to the controlled group. In an exploratory 1-year clinical trial evaluating the use of 50 mg, 100 mg, and 200 mg of SIMPONI in 309 patients with severe persistent asthma, 6 patients developed malignancies other than NMSC in the SIMPONI groups compared to none in the control group. Three of the 6 patients were in the 200 mg SIMPONI group.
Congestive Heart Failure
Cases of worsening congestive heart failure (CHF) and new onset CHF have been reported with TNF blockers, including SIMPONI. Some cases had a fatal outcome. In several exploratory trials of other TNF blockers in the treatment of CHF, there were greater proportions of TNF-blocker-treated patients who had CHF exacerbations requiring hospitalization or increased mortality. SIMPONI has not been studied in patients with a history of CHF and SIMPONI should be used with caution in patients with CHF. If a decision is made to administer SIMPONI to patients with CHF, these patients should be closely monitored during therapy, and SIMPONI should be discontinued if new or worsening symptoms of CHF appear.
Demyelinating Disorders
Use of TNF blockers, of which SIMPONI is a member, has been associated with rare cases of new onset or exacerbation of central nervous system (CNS) demyelinating disorders, including multiple sclerosis (MS) and peripheral demyelinating disorders, including Guillain-Barré syndrome. Cases of central demyelination, MS, optic neuritis, and peripheral demyelinating polyneuropathy have rarely been reported in patients treated with SIMPONI [see Adverse Reactions (6.1) ] . Prescribers should exercise caution in considering the use of TNF blockers, including SIMPONI, in patients with central or peripheral nervous system demyelinating disorders. Discontinuation of SIMPONI should be considered if these disorders develop.
Hepatitis B Virus Reactivation
The use of TNF blockers including SIMPONI has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic hepatitis B carriers (i.e., surface antigen positive). In some instances, HBV reactivation occurring in conjunction with TNF blocker therapy has been fatal. The majority of these reports have occurred in patients who received concomitant immunosuppressants.
All patients should be tested for HBV infection before initiating TNF-blocker therapy. For patients who test positive for hepatitis B surface antigen, consultation with a physician with expertise in the treatment of hepatitis B is recommended before initiating TNF-blocker therapy. The risks and benefits of treatment should be considered prior to prescribing TNF blockers, including SIMPONI, to patients who are carriers of HBV. Adequate data are not available on whether antiviral therapy can reduce the risk of HBV reactivation in HBV carriers who are treated with TNF blockers. Patients who are carriers of HBV and require treatment with TNF blockers should be closely monitored for clinical and laboratory signs of active HBV infection throughout therapy and for several months following termination of therapy.
In patients who develop HBV reactivation, TNF blockers should be stopped and antiviral therapy with appropriate supportive treatment should be initiated. The safety of resuming TNF blockers after HBV reactivation has been controlled is not known. Therefore, prescribers should exercise caution when considering resumption of TNF blockers in this situation and monitor patients closely.
Autoimmunity
Treatment with TNF blockers, including SIMPONI, may result in the formation of antinuclear antibodies (ANA) and, rarely, in the development of a lupus-like syndrome [see Adverse Reactions (6.1) ] . If a patient develops symptoms suggestive of a lupus-like syndrome following treatment with SIMPONI, treatment should be discontinued.
Use with Abatacept
In controlled trials, the concurrent administration of another TNF blocker and abatacept was associated with a greater proportion of serious infections than the use of a TNF blocker alone; and the combination therapy, compared to the use of a TNF blocker alone, has not demonstrated improved clinical benefit in the treatment of RA. Therefore, the combination of TNF blockers, including SIMPONI, and abatacept is not recommended [see Drug Interactions (7.2) ] .
Use with Anakinra
Concurrent administration of anakinra (an interleukin-1 antagonist) and another TNF blocker was associated with a greater portion of serious infections and neutropenia and no additional benefits compared with the TNF-blocker alone. Therefore, the combination of anakinra with TNF blockers, including SIMPONI, is not recommended [see Drug Interactions (7.2) ] .
Switching Between Biological Disease-Modifying Antirheumatic Drugs
Care should be taken when switching from one biological product to another biological product since overlapping biological activity may further increase the risk of infection.
Hematologic Cytopenias
There have been reports of pancytopenia, leukopenia, neutropenia, agranulocytosis, aplastic anemia, and thrombocytopenia in patients receiving golimumab. Caution should be exercised when using TNF blockers, including SIMPONI, in patients who have or have had significant cytopenias.
Vaccinations/Therapeutic Infectious Agents
Live Vaccines
Patients treated with SIMPONI may receive vaccinations, except for live vaccines. In patients receiving anti-TNF therapy, limited data are available on the response to live vaccination, or on the secondary transmission of infection by live vaccines. Use of live vaccines could result in clinical infections, including disseminated infections.
If possible, it is recommended that prior to initiating therapy with SIMPONI, pediatric patients be brought up to date with all immunizations in agreement with current immunization guidelines.
Therapeutic Infectious Agents
Other uses of therapeutic infectious agents such as live attenuated bacteria (e.g., BCG bladder instillation for the treatment of cancer) could result in clinical infections, including disseminated infections. It is recommended that therapeutic infectious agents not be given concurrently with SIMPONI.
Non-live Vaccines
In the Phase 3 PsA trial, after pneumococcal vaccination, a similar proportion of SIMPONI-treated and placebo-treated patients were able to mount an adequate immune response of at least a 2-fold increase in antibody titers to pneumococcal polysaccharide vaccine. In both SIMPONI-treated and placebo-treated patients, the proportions of patients with response to pneumococcal vaccine were lower among patients receiving MTX compared with patients not receiving MTX. The data suggest that SIMPONI does not suppress the humoral immune response to the pneumococcal vaccine.
Hypersensitivity Reactions
In postmarketing experience, serious systemic hypersensitivity reactions (including anaphylactic reaction) have been reported following SIMPONI administration. Some of these reactions occurred after the first administration of SIMPONI. If an anaphylactic or other serious allergic reaction occurs, administration of SIMPONI should be discontinued immediately and appropriate therapy instituted.
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions (5.1) ]
- Malignancies [see Warnings and Precautions (5.2) ]
- Congestive Heart Failure [see Warnings and Precautions (5.3) ]
- Demyelinating Disorders [see Warnings and Precautions (5.4) ]
- Hepatitis B Reactivation [see Warnings and Precautions (5.5) ]
- Autoimmunity [see Warnings and Precautions (5.6) ]
- Hematologic Cytopenias [see Warnings and Precautions (5.10) ]
- Hypersensitivity Reactions [see Warnings and Precautions (5.12) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety data described below are based on:
- Five pooled, randomized, double-blind, controlled Phase 3 trials in patients with RA, PsA, and AS (Trials RA-1, RA-2, RA-3, PsA, and AS) [see Clinical Studies (14.1 , 14.2 , and 14.3) ] . These 5 trials included 639 control-treated patients and 1659 SIMPONI-treated patients including 1089 with RA, 292 with PsA, and 278 with AS.
- Three pooled, randomized, double-blind, controlled Phase 2/3 in 1233 SIMPONI-treated adult patients with UC (Trials UC-1, UC-2, and UC-3 (NCT03596645)) [see Clinical Studies (14.4) ] .
- An open-label trial in 69 SIMPONI-treated pediatric patients weighing at least 15 kg with UC (Trial UC-3) (NCT03596645) [see Clinical Studies (14.5) ] .
The proportion of adult patients who discontinued treatment due to adverse reactions in the controlled Phase 3 trials through Week 16 in RA, PsA and AS was 2% for SIMPONI-treated patients and 3% for placebo-treated patients. The most common adverse reactions leading to discontinuation of SIMPONI in the controlled Phase 3 trials in RA, PsA and AS through Week 16 were sepsis (0.2%), alanine aminotransferase increased (0.2%), and aspartate aminotransferase increased (0.2%). The most common adverse drug reactions leading to discontinuation through Week 60 of the UC trials in adult patients who received SIMPONI induction and 100 mg during maintenance compared with patients who received SIMPONI induction and placebo during maintenance were tuberculosis (0.3% vs. 0.6%) and anemia (0.3% vs. 0%), respectively.
Upper respiratory tract infection and nasopharyngitis were the most common adverse reactions reported in the combined Phase 3 RA, PsA and AS trials in adults through Week 16, occurring in 7% and 6% of SIMPONI-treated patients as compared with 6% and 5% of control-treated patients, respectively.
Infections
In adult controlled Phase 3 trials through Week 16 in RA, PsA, and AS, infections were observed in 28% of SIMPONI-treated patients compared to 25% of control-treated patients. For serious infections, see the Warnings and Precautions section [see Warnings and Precautions (5.1) ] . In Trial UC-1, the rates of infections were similar in SIMPONI 200/100 mg-treated patients and placebo-treated patients, or approximately 12%. Through Week 60, the incidence per patient year of infections was similar in patients who received SIMPONI induction and 100 mg during maintenance compared with patients who received SIMPONI induction and placebo during the maintenance period Trial UC-2.
Demyelinating Disorders
In Trial UC-1 of SIMPONI induction through Week 6, no cases of demyelination were observed in SIMPONI 200/100 mg-treated patients or placebo-treated patients. Through Week 60 in Trial UC-2, there were no cases of demyelination in the SIMPONI 100 mg group during maintenance. One case of CNS demyelination was observed in the placebo maintenance group in a patient who received SIMPONI 400/200 mg during induction.
Liver Enzyme Elevations
There have been reports of severe hepatic reactions including acute liver failure in patients receiving TNF blockers. In adult controlled Phase 3 trials of SIMPONI in patients with RA, PsA, and AS through Week 16, ALT elevations ≥ 5 × ULN occurred in 0.2% of control-treated patients and 0.7% of SIMPONI-treated patients and ALT elevations ≥ 3 × ULN occurred in 2% of control-treated patients and 2% of SIMPONI-treated patients. Since many of the patients in the Phase 3 trials for RA, PsA, and AS were also taking medications that cause liver enzyme elevations (e.g., NSAIDs, MTX), the relationship between SIMPONI and liver enzyme elevation is not clear.
In Trials UC-1, UC-2, and UC-3, the incidence of ALT elevations ≥ 5 × ULN was similar in SIMPONI-treated patients and placebo-treated patients, or approximately 1%, with an average duration of follow-up of 46 weeks and 18 weeks, respectively. ALT elevations ≥ 3 × ULN occurred in 2.0% of SIMPONI-treated patients compared with 1.5% of placebo-treated patients with an average duration of follow-up of 46 weeks and 18 weeks, respectively.
Autoimmune Disorders and Autoantibodies
In the adult controlled Phase 3 trials in patients with RA, PsA, and AS through Week 14, there was no association of SIMPONI treatment and the development of newly positive anti-dsDNA antibodies. In Phase 3 trials in RA, PsA, and AS through 1 year of follow-up, 4.0% of SIMPONI-treated patients and 2.6% of control patients were newly antinuclear antibody (ANA)-positive (at titers of 1:160 or greater). The frequency of anti-dsDNA antibodies at 1 year of follow-up was uncommon in patients who were anti-dsDNA negative at baseline. Through Week 60 of the UC trials (Trials UC-1, UC-2, and UC-3), 3.5% of patients who received SIMPONI induction and 100 mg during maintenance were newly ANA-positive (at titers of 1:160 or greater) compared with 3.5% of patients who received SIMPONI induction and placebo during the maintenance period in Trial UC-2. The frequency of anti-dsDNA antibodies at 1 year of follow-up in patients who were anti-dsDNA negative at baseline was 0.5% in patients receiving SIMPONI induction and 100 mg during maintenance compared with 0% in patients who received SIMPONI induction and placebo during maintenance [see Warnings and Precautions (5.6) ] .
Injection Site Reactions
In adult controlled Phase 3 trials through Week 16 in RA, PsA and AS, 6% of SIMPONI-treated patients had injection site reactions compared with 2% of control-treated patients. The majority of the injection site reactions were mild and the most frequent manifestation was injection site erythema.
In Trial UC-1, 3.4% of SIMPONI-treated patients had injection site reactions compared with 1.5% in control-treated patients. The majority of the injection site reactions were mild and moderate and the most frequent manifestation was injection site erythema.
In adult controlled Phase 2 and 3 trials in RA, PsA, AS, no patients treated with SIMPONI developed anaphylactic reactions.
Other Adverse Reactions
Table 2 summarizes the adverse drug reactions that occurred at a rate of at least 1% in the SIMPONI ± DMARD group and with a higher incidence than in the placebo ± DMARD group during the controlled period of the 5 pooled Phase 3 trials through Week 16 in adult patients with RA, PsA, and AS.
| SIMPONI ± DMARDs | Placebo ± DMARDs | |
|---|---|---|
| Patients treated | 1659 | 639 |
| Adverse Reaction | ||
| Infections and infestations | ||
| Upper respiratory tract infection (nasopharyngitis, pharyngitis, laryngitis, and rhinitis) | 16% | 13% |
| Viral infections (such as influenza and herpes) | 5% | 3% |
| Bronchitis | 2% | 1% |
| Superficial fungal infections | 2% | 1% |
| Sinusitis | 2% | 1% |
| General disorders and administration site conditions | ||
| Injection site reaction (injection site erythema, urticaria, induration, pain, bruising, pruritus, irritation, paresthesia) | 6% | 2% |
| Investigations | ||
| Alanine aminotransferase increased | 4% | 3% |
| Aspartate aminotransferase increased | 3% | 2% |
| Vascular disorders | ||
| Hypertension | 3% | 2% |
| Nervous system disorders | ||
| Dizziness | 2% | 1% |
| Paresthesia | 2% | 1% |
| Gastrointestinal disorders | ||
| Constipation | 1% | <1% |
Less Common Clinical Trial Adverse Drug Reactions
Adverse drug reactions that occurred <1% in adult SIMPONI-treated patients in the RA, PsA and AS clinical trials that do not appear in the Warnings and Precautions section included the following events listed by system organ class:
Infections and infestations: Septic shock, atypical mycobacterial infection, pyelonephritis, arthritis bacterial, bursitis infective
Neoplasms benign, malignant and unspecified: Leukemia
Skin and subcutaneous tissue disorders: Psoriasis (new onset or worsening, palmar/plantar and pustular), vasculitis (cutaneous)
Vascular disorders: Vasculitis (systemic)
Adults and Pediatric Patients Weighing at least 15 kg with Ulcerative Colitis
In general, adverse reactions reported in adult patients with UC in Trials UC-1, UC-2, and UC-3 were similar to those reported in clinical trials of patients with RA, PsA, and AS. Adverse reactions reported in the clinical trial of pediatric patients weighing at least 15 kg with UC were also similar to those reported in clinical trials of adults with UC and the other indicated populations. Additional adverse reactions reported in at least 10% of pediatric patients in the trial were headache (17%) and pyrexia (10%).
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of golimumab. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to SIMPONI exposure.
Immune system disorders: Serious systemic hypersensitivity reactions (including anaphylactic reaction) [see Warnings and Precautions (5.12) ] , sarcoidosis
Neoplasms benign, malignant and unspecified: Melanoma, Merkel cell carcinoma [see Warnings and Precautions (5.2) ]
Respiratory, thoracic and mediastinal disorders: Interstitial lung disease
Skin and subcutaneous tissue disorders : Skin exfoliation, lichenoid reactions, rash, bullous skin reactions
DRUG INTERACTIONS
Methotrexate
For the treatment of RA, SIMPONI should be used with methotrexate (MTX) [see Clinical Studies (14.1) ] . Since the presence or absence of concomitant MTX did not appear to influence the efficacy or safety of SIMPONI in the treatment of PsA or AS, SIMPONI can be used with or without MTX in the treatment of PsA and AS [see Clinical Studies (14.2 , 14.3) and Clinical Pharmacology (12.3) ] .
Biological Products for RA, PsA, and/or AS
An increased risk of serious infections has been seen in clinical RA trials of other TNF blockers used in combination with anakinra or abatacept, with no added benefit; therefore, use of SIMPONI with abatacept or anakinra is not recommended [see Warnings and Precautions (5.7 , 5.8) ] . A higher rate of serious infections has also been observed in RA patients treated with rituximab who received subsequent treatment with a TNF blocker. The concomitant use of SIMPONI with biologics approved to treat RA, PsA, or AS is not recommended because of the possibility of an increased risk of infection.
Live Vaccines/Therapeutic Infectious Agents
Live vaccines should not be given concurrently with SIMPONI [see Warnings and Precautions (5.11) ] .
Therapeutic infectious agents should not be given concurrently with SIMPONI [see Warnings and Precautions (5.11) ] .
Infants born to women treated with SIMPONI during their pregnancy may be at increased risk of infection for up to 6 months. Administration of live vaccines to infants exposed to SIMPONI in utero is not recommended for 6 months following the mother's last SIMPONI injection during pregnancy [see Use in Specific Populations (8.1) ] .
Cytochrome P450 Substrates
The formation of CYP450 enzymes may be suppressed by increased levels of cytokines (e.g., TNFα) during chronic inflammation. Therefore, it is expected that for a molecule that antagonizes cytokine activity, such as golimumab, the formation of CYP450 enzymes could be normalized. Upon initiation or discontinuation of SIMPONI in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended and the individual dose of the drug product may be adjusted as needed.
DESCRIPTION
Golimumab is a human IgG1κ monoclonal antibody specific for human tumor necrosis factor alpha (TNFα) that exhibits multiple glycoforms with molecular masses of approximately 150 to 151 kilodaltons. Golimumab was created using genetically engineered mice immunized with human TNF, resulting in an antibody with human-derived antibody variable and constant regions. Golimumab is produced by a recombinant cell line cultured by continuous perfusion and is purified by a series of steps that includes measures to inactivate and remove viruses.
SIMPONI ® (golimumab) Injection is a preservative-free, sterile, clear to slightly opalescent, colorless to light yellow solution of the golimumab antibody supplied in a single-dose prefilled syringe (with a passive needle safety guard) or a single-dose prefilled autoinjector.
Each 0.5 mL prefilled syringe and autoinjector contains 50 mg golimumab, L-histidine and L-histidine monohydrochloride monohydrate (0.44 mg), polysorbate 80 (0.08 mg), sorbitol (20.5 mg) and Water for Injection. Each 1 mL prefilled syringe and autoinjector contains 100 mg golimumab, L-histidine and L-histidine monohydrochloride monohydrate (0.87 mg), polysorbate 80 (0.15 mg), sorbitol (41.0 mg) and Water for Injection. The pH is approximately 5.5.
CLINICAL PHARMACOLOGY
Mechanism of Action
Golimumab is a human monoclonal antibody that binds to both the soluble and transmembrane bioactive forms of human TNFα. This interaction prevents the binding of TNFα to its receptors, thereby inhibiting the biological activity of TNFα (a cytokine protein). There was no evidence of the golimumab antibody binding to other TNF superfamily ligands; in particular, the golimumab antibody did not bind or neutralize human lymphotoxin. Golimumab did not lyse human monocytes expressing transmembrane TNF in the presence of complement or effector cells.
Elevated TNFα levels in the blood, synovium, and joints have been implicated in the pathophysiology of several chronic inflammatory diseases such as rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. TNFα is an important mediator of the articular inflammation that is characteristic of these diseases. The exact mechanism by which golimumab treats ulcerative colitis is unknown. Golimumab modulated the in vitro biological effects mediated by TNF in several bioassays, including the expression of adhesion proteins responsible for leukocyte infiltration (E-selectin, ICAM-1 and VCAM-1) and the secretion of proinflammatory cytokines (IL-6, IL-8, G-CSF and GM-CSF).
Pharmacodynamics
In clinical trials, decreases in C-reactive protein (CRP), interleukin (IL)-6, matrix metalloproteinase-3 (MMP-3), intercellular adhesion molecule (ICAM)-1 and vascular endothelial growth factor (VEGF) were observed following SIMPONI administration in patients with RA, PsA, and AS.
Pharmacokinetics
Absorption
Following subcutaneous administration of SIMPONI to healthy subjects and patients with active RA, the median time to reach maximum serum concentrations (T max ) ranged from 2 to 6 days. A subcutaneous injection of 50 mg SIMPONI to healthy subjects produced a mean ± standard deviation maximum serum concentration (C max ) of 3.2 ± 1.4 mcg/mL.
By cross-trial comparisons of mean AUC inf values following an IV or subcutaneous administration of SIMPONI, the absolute bioavailability of subcutaneous SIMPONI was estimated to be approximately 53%.
Distribution
Following a single IV administration over the dose range of 0.1 to 10.0 mg/kg in patients with active RA, mean volume of distribution ranged from 58 to 126 mL/kg. The volume of distribution for golimumab indicates that golimumab is distributed primarily in the circulatory system with limited extravascular distribution.
Metabolism
The exact metabolic pathway of golimumab is unknown.
Elimination
Following a single IV administration over the dose range of 0.1 to 10.0 mg/kg in patients with active RA, mean systemic clearance of SIMPONI was estimated to be 4.9 to 6.7 mL/day/kg.
Median terminal half-life values were estimated to be approximately 2 weeks in healthy subjects and patients with active RA, PsA or AS.
Population PK analyses indicated that concomitant use of NSAIDs, oral corticosteroids, or sulfasalazine did not influence the apparent clearance of golimumab.
Patients who developed anti-golimumab antibodies generally had lower steady-state serum trough concentrations of golimumab.
Dose Linearity
Golimumab exhibited dose-proportional pharmacokinetics (PK) in patients with active RA over the dose range of 0.1 to 10 mg/kg following a single intravenous (IV) dose. Following a single SC dose in healthy subjects, dose proportional pharmacokinetics were also observed over a dose range of 50 mg to 400 mg.
Single Dose Versus Multiple Doses
When 50 mg SIMPONI was administered subcutaneous to patients with RA, PsA, or AS every 4 weeks, serum concentrations appeared to reach steady state by Week 12. With concomitant use of methotrexate (MTX), treatment with 50 mg SIMPONI subcutaneous every 4 weeks resulted in a mean steady-state trough serum concentration of approximately 0.4–0.6 mcg/mL in patients with active RA, approximately 0.5 mcg/mL in patients with active PsA, and approximately 0.8 mcg/mL in patients with active AS. Patients with RA, PsA, and AS treated with SIMPONI 50 mg and MTX had approximately 52%, 36% and 21% higher mean steady-state trough concentrations of golimumab, respectively compared with those treated with SIMPONI 50 mg without MTX. The presence of MTX also decreased anti-golimumab antibody incidence from 7% to 2% [see Adverse Reactions (6.1) ] . For RA, SIMPONI should be used with MTX. In the PsA and AS trials, the presence or absence of concomitant MTX did not appear to influence clinical efficacy and safety parameters [see Drug Interactions (7.1) and Clinical Studies (14.1) ] .
When induction doses of 200 mg and 100 mg SIMPONI at week 0 and 2, respectively, followed by maintenance doses of 100 mg SIMPONI every 4 weeks were administered subcutaneously in patients with UC, serum golimumab concentrations reached steady-state by week 8 after the first maintenance dose. Treatment with 100 mg SIMPONI subcutaneous every 4 weeks during maintenance resulted in a mean steady-state trough serum concentration of approximately 1.8 ± 1.1 mcg/mL.
Effect of Weight on Pharmacokinetics in Adults
Higher apparent clearance of golimumab was associated with increasing weight. Treatment with the recommended maintenance dose regimen of SIMPONI 100 mg in adult patients with UC did not result in meaningful differences in clinical efficacy among different weight groups. Across the PsA and AS populations, no meaningful differences in clinical efficacy were observed among the subgroups by weight quartile. The RA trial in MTX-experienced and TNF-blocker-naïve patients (Trial RA-2) did show evidence of a reduction in clinical efficacy with increasing body weight, but this effect was observed for both tested doses of SIMPONI (50 mg and 100 mg). There is no need to adjust the dosage of SIMPONI in adult patients based on body weight.
Specific Populations
In adults, population PK analyses suggested no PK differences between male and female patients after body-weight adjustment in the RA, PsA and UC trials. In the AS trial, female patients showed 13% higher apparent clearance than male patients after body-weight adjustment. Subgroup analysis based on gender showed that both female and male patients achieved clinically significant response at the proposed clinical dose. Dosage adjustment based on gender is not needed.
Population PK analyses indicated that PK parameters of golimumab were not influenced by age in adult patients. Patients with age ≥ 65 years had apparent clearance of golimumab similar to patients with age < 65 years. No ethnicity-related PK differences were observed between Caucasians and Asians, and there were too few patients of other races to assess for PK differences.
No formal trial of the effect of renal or hepatic impairment on the PK of golimumab was conducted.
Pediatric Patients Weighing at Least 15 kg with Ulcerative Colitis
The pharmacokinetics of golimumab were studied in pediatric patients with UC aged 4 to 17 years. The pharmacokinetics of golimumab are predicted to be similar between adult patients and pediatric patients with UC. There are no expected clinically significant differences in golimumab systemic exposure between adult patients and pediatric patients weighing 15 kg and above who receive the recommended dosage [see Dosage and Administration (2.3) ] .
Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of golimumab or of other golimumab products.
Results from the EIA Method
Using an enzyme immunoassay (EIA method), antibodies to golimumab were detected in 57 (4%) of SIMPONI-treated adult patients across the Phase 3 RA, PsA and AS trials through Week 24. Similar rates were observed in each of the 3 indications. Patients who received SIMPONI with concomitant MTX had a lower proportion of antibodies to golimumab than patients who received SIMPONI without MTX (approximately 2% vs. 7%, respectively).
With the EIA method, the presence of serum concentrations of golimumab can interfere with the detection of antibodies to golimumab leading to inconclusive results. In adult UC trials, 34 (3%), 341 (28%) and 823 (69%) of SIMPONI-treated patients were positive, negative and inconclusive for antibodies to golimumab, respectively. Treatment with concomitant immunomodulators (AZA, 6-MP or MTX) resulted in a lower proportion of patients with antibodies to golimumab than patients receiving SIMPONI without immunomodulators (2% vs. 4%, respectively).
Of the adult patients with a positive antibody response to golimumab in the Phase 2 and 3 trials, most were determined to have neutralizing antibodies to golimumab as measured by a cell-based functional assay.
Results from the Drug-Tolerant EIA Method
A drug-tolerant enzyme immunoassay (drug-tolerant EIA) method for detecting antibodies to golimumab was developed and validated, which eliminated the inconclusive category as reported above. This method is approximately 16-fold more sensitive than the original EIA method with less interference from golimumab in serum.
Based on the drug-tolerant EIA method, 246 (23%) of SIMPONI-treated patients across the Phase 3 RA, PsA and AS trials in adults, antibodies to golimumab were detected in 59 (16%), 106 (28%), and 81 (24%) patients, respectively. Treatment with concomitant MTX resulted in a lower proportion of patients with antibodies to golimumab than in patients receiving SIMPONI without MTX in RA patients (7% vs. 35%), in PsA patients (18% vs. 38%) and in AS patients (6% vs. 29%). A trend of decreasing drug concentrations with increasing antibody titers was observed. While an overall decrease in clinical efficacy for ADA positive patients compared with ADA negative patients was not observed in patients with RA (ACR 20: 75% vs. 75%), PsA (ACR 20: 72% vs. 66%) and AS (ASAS 20: 57% vs. 65%), higher titer antibodies may be associated with diminished efficacy.
In UC trials in adults, 254 (21%) of SIMPONI-treated patients were positive for antibodies to golimumab through week 54 while the remaining 941 (79%) patients were negative. Treatment with concomitant immunomodulators (AZA, 6-MP or MTX) in the UC trials resulted in a lower proportion of patients with antibodies to golimumab than in patients receiving SIMPONI without immunomodulators (12% vs. 26%). There is a trend of decreasing drug concentrations with increasing antibody titers. Although the development of antibodies to golimumab did not preclude clinical response, a trend toward decreased efficacy in ADA positive patients was observed compared to ADA negative patients in the UC trials (clinical response 38% vs. 53%).
In the pediatric UC trial, 22% (15/69) of SIMPONI-treated patients developed ADA to golimumab through the final safety visit using the drug-tolerant EIA method. Of the 15 subjects who tested positive for ADA, 20% (3/15) tested positive for neutralizing antibodies. The incidence of antibodies to golimumab was comparable between pediatric and adult patients with UC. Pediatric UC patients who developed antibodies to golimumab generally had lower trough steady-state serum concentrations of golimumab. While there is insufficient data to assess whether the observed ADA-associated pharmacokinetic changes reduce effectiveness, a trend toward decreased efficacy in ADA positive patients was observed compared to ADA negative patients in the pediatric UC trial.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies of golimumab have not been conducted to evaluate its carcinogenic potential. Mutagenicity studies have not been conducted with golimumab. A fertility study conducted in mice using an analogous anti-mouse TNFα antibody administered by the intravenous route at doses up to 40 mg/kg once per week showed no impairment of fertility.
CLINICAL STUDIES
Rheumatoid Arthritis
The efficacy and safety of SIMPONI were evaluated in 3 multicenter, randomized, double-blind, controlled trials (Trials RA-1, RA-2, and RA-3) in 1542 patients ≥ 18 years of age with moderately to severely active RA, diagnosed according to the American College of Rheumatology (ACR) criteria, for at least 3 months prior to administration of trial agent. Patients were required to have at least 4 swollen and 4 tender joints. SIMPONI was administered subcutaneously at doses of 50 mg or 100 mg every 4 weeks. Double-blinded controlled efficacy data were collected and analyzed through Week 24. Patients were allowed to continue stable doses of concomitant low dose corticosteroids (equivalent to ≤ 10 mg of prednisone a day) and/or NSAIDs and patients may have received oral MTX during the trials.
Trial RA-1 evaluated 445 patients who were previously treated (at least 8 to 12 weeks prior to administration of trial agent) with one or more doses of a biologic TNF blocker without a serious adverse reaction. Patients may have discontinued the biologic TNF blocker for a variety of reasons. Patients were randomized to receive placebo (N=150), SIMPONI 50 mg (N=147), or SIMPONI 100 mg (N=148). Patients were allowed to continue stable doses of concomitant MTX, sulfasalazine (SSZ), and/or hydroxychloroquine (HCQ) during the trial. The use of other DMARDs including cytotoxic agents or other biologics was prohibited.
Trial RA-2 evaluated 444 patients who had active RA despite a stable dose of at least 15 mg/week of MTX and who had not been previously treated with a biologic TNF blocker. Patients were randomized to receive background MTX (N=133), SIMPONI 50 mg + background MTX (N=89), SIMPONI 100 mg + background MTX (N=89), or SIMPONI 100 mg monotherapy (N=133). The use of other DMARDs including SSZ, HCQ, cytotoxic agents, or other biologics was prohibited.
Trial RA-3 evaluated 637 patients with active RA who were MTX naïve and had not previously been treated with a biologic TNF blocker. Patients were randomized to receive MTX (N=160), SIMPONI 50 mg + MTX (N=159), SIMPONI 100 mg + MTX (N=159), or SIMPONI 100 mg monotherapy (N=159). For patients receiving MTX, MTX was administered at a dose of 10 mg/week beginning at Week 0 and increased to 20 mg/week by Week 8. The use of other DMARDs including SSZ, HCQ, cytotoxic agents, or other biologics was prohibited.
The primary endpoint in Trial RA-1 and Trial RA-2 was the percentage of patients achieving an ACR 20 response at Week 14 and the primary endpoint in Trial RA-3 was the percentage of patients achieving an ACR 50 response at Week 24.
In Trials RA-1, RA-2, and RA-3, the median duration of RA disease was 9.4, 5.7, and 1.2 years and 99%, 75%, and 54% of the patients used at least one DMARD in the past, respectively. Approximately 77% and 57% of patients received concomitant NSAIDs and low dose corticosteroids, respectively, in the 3 pooled RA trials.
Clinical Response
In the 3 RA trials, a greater percentage of patients treated with the combination of SIMPONI and MTX achieved ACR responses at Week 14 (Trials RA-1 and RA-2) and Week 24 (Studies RA-1, RA-2, and RA-3) versus patients treated with the MTX alone. There was no clear evidence of improved ACR response with the higher SIMPONI dose group (100 mg) compared to the lower SIMPONI dose group (50 mg). In Trials RA-2 and RA-3, the SIMPONI monotherapy groups were not statistically different from the MTX monotherapy groups in ACR responses. Table 3 shows the proportion of patients with the ACR response for the SIMPONI 50 mg and control groups in Trials RA-1, RA-2, and RA-3. In the subset of patients who received SIMPONI in combination with MTX in Trial RA-1, the proportion of patients achieving ACR 20, 50 and 70 responses at Week 14 were 40%, 18%, and 12%, respectively, in the SIMPONI 50 mg + MTX group (N=101) compared with 17%, 6%, and 2%, respectively, in the placebo + MTX group (N=103). Table 4 shows the percent improvement in the components of the ACR response criteria for the SIMPONI 50 mg + MTX and MTX groups in Trial RA-2. The percentage of patients achieving ACR 20 responses by visit for Trial RA-2 is shown in Figure 1. ACR 20 responses were observed in 38% of patients in the SIMPONI 50 mg + MTX group at the first assessment (Week 4) after the initial SIMPONI administration.
| Trial RA-1 Active RA previously treated with one or more doses of TNF blockers | Trial RA-2 Active RA, despite MTX | Trial RA-3 Active RA, MTX Naïve | ||||
|---|---|---|---|---|---|---|
| Placebo ± DMARDs DMARDs in Trial RA-1 included MTX, HCQ, and/or SSZ (about 68%, 8%, and 5% of patients received MTX, HCQ, and SSZ, respectively). | SIMPONI 50 mg ± DMARDs | Background MTX | SIMPONI 50 mg + Background MTX | MTX | SIMPONI 50 mg + MTX | |
| N N reflects randomized patients. | 150 | 147 | 133 | 89 | 160 | 159 |
| ACR 20 | ||||||
| Week 14 | 18% | 35% | 33% | 55% | NA NA = Not applicable, as data was not collected at Week 14 in Trial RA-3. | NA |
| Week 24 | 16% | 31% | 28% | 60% | 49% | 62% |
| ACR 50 | ||||||
| Week 14 | 7% | 15% | 10% | 35% | NA | NA |
| Week 24 | 4% | 16% | 14% | 37% | 29% | 40% |
| ACR 70 | ||||||
| Week 14 | 2% | 10% | 4% | 13% | NA | NA |
| Week 24 | 2% | 9% | 5% | 20% | 16% | 24% Not significantly different from MTX monotherapy. |
| Background MTX | SIMPONI 50 mg + Background MTX | |
|---|---|---|
| Note: Baseline values are medians. | ||
| N N reflects randomized patients; actual number of patients evaluable for each endpoint may vary. | 133 | 89 |
| Number of swollen joints (0–66) | ||
| Baseline | 12 | 13 |
| Week 14 | 38% | 62% |
| Number of tender joints (0–68) | ||
| Baseline | 21 | 26 |
| Week 14 | 30% | 60% |
| Patient's assessment of pain (0–10) | ||
| Baseline | 5.7 | 6.1 |
| Week 14 | 18% | 55% |
| Patient's global assessment of disease activity (0–10) | ||
| Baseline | 5.3 | 6.0 |
| Week 14 | 15% | 45% |
| Physician's global assessment of disease activity (0–10) | ||
| Baseline | 5.7 | 6.1 |
| Week 14 | 35% | 55% |
| HAQ score (0–3) | ||
| Baseline | 1.25 | 1.38 |
| Week 14 | 10% | 29% |
| CRP (mg/dL) | ||
| Baseline | 0.8 | 1.0 |
| Week 14 | 2% | 44% |
| Figure 1: Trial RA-2 – Percentage of Patients Achieving ACR 20 Response by Visit: Randomized Patients The same patients may not have responded at each timepoint. |
 |
Physical Function Response in Patients with RA
In Trials RA-1 and RA-2, the SIMPONI 50 mg groups demonstrated a greater improvement compared to the control groups in the change in mean Health Assessment Questionnaire Disability Index (HAQ-DI) score from baseline to Week 24: 0.23 vs. 0.03 in RA-1, 0.47 vs. 0.13 in RA-2, respectively. Also in Trials RA-1 and RA-2, the SIMPONI 50 mg groups compared to the control groups had a greater proportion of HAQ responders (change from baseline > 0.22) at Week 24: 43% vs. 27%, 65% vs. 35%, respectively.
Psoriatic Arthritis
The safety and efficacy of SIMPONI were evaluated in a multicenter, randomized, double-blind, placebo-controlled trial in 405 adult patients with moderately to severely active PsA (≥ 3 swollen joints and ≥ 3 tender joints) despite NSAID or DMARD therapy (Trial PsA). Patients in this trial had a diagnosis of PsA for at least 6 months with a qualifying psoriatic skin lesion of at least 2 cm in diameter. Previous treatment with a biologic TNF blocker was not allowed. Patients were randomly assigned to placebo (N=113), SIMPONI 50 mg (N=146), or SIMPONI 100 mg (N=146) given subcutaneously every 4 weeks. Patients were allowed to receive stable doses of concomitant MTX (≤ 25 mg/week), low dose oral corticosteroids (equivalent to ≤ 10 mg of prednisone a day), and/or NSAIDs during the trial. The use of other DMARDs including SSZ, HCQ, cytotoxic agents, or other biologics was prohibited. The primary endpoint was the percentage of patients achieving ACR 20 response at Week 14. Placebo-controlled efficacy data were collected and analyzed through Week 24.
Patients with each subtype of PsA were enrolled, including polyarticular arthritis with no rheumatoid nodules (43%), asymmetric peripheral arthritis (30%), distal interphalangeal (DIP) joint arthritis (15%), spondylitis with peripheral arthritis (11%), and arthritis mutilans (1%). The median duration of PsA disease was 5.1 years, 78% of patients received at least one DMARD in the past, and approximately 48% of patients received MTX, and 16% received low dose oral steroids.
Clinical Response in Patients with PsA
SIMPONI ± MTX, compared with placebo ± MTX, resulted in significant improvement in signs and symptoms as demonstrated by the proportion of patients with an ACR 20 response at Week 14 in Trial PsA (see Table 5 ). There was no clear evidence of improved ACR response with the higher SIMPONI dose group (100 mg) compared to the lower SIMPONI dose group (50 mg). ACR responses observed in the SIMPONI-treated groups were similar in patients receiving and not receiving concomitant MTX. Similar ACR 20 responses at Week 14 were observed in patients with different PsA subtypes. However, the number of patients with arthritis mutilans was too small to allow meaningful assessment. SIMPONI 50 mg treatment also resulted in significantly greater improvement compared with placebo for each ACR component in Trial PsA (Table 6). Treatment with SIMPONI resulted in improvement in enthesitis and skin manifestations in patients with PsA. However, the safety and efficacy of SIMPONI in the treatment of patients with plaque psoriasis has not been established.
The percentage of patients achieving ACR 20 responses by visit for Trial PsA is shown in Figure 2. ACR 20 responses were observed in 31% of patients in the SIMPONI 50 mg + MTX group at the first assessment (Week 4) after the initial SIMPONI administration.
| Placebo ± MTX In Trial PsA, about 48%, 16%, and 72% of the patients received stable doses of MTX (≤ 25 mg/week), low dose corticosteroids (equivalent to ≤ 10 mg of prednisone a day), and NSAIDs, respectively. | SIMPONI 50 mg ± MTX | |
|---|---|---|
| Bold text indicates primary endpoint. | ||
| N N reflects randomized patients. | 113 | 146 |
| ACR 20 | ||
| Week 14 | 9% | 51% |
| Week 24 | 12% | 52% |
| ACR 50 | ||
| Week 14 | 2% | 30% |
| Week 24 | 4% | 32% |
| ACR 70 | ||
| Week 14 | 1% | 12% |
| Week 24 | 1% | 19% |
| Placebo ± MTX In Trial PsA, about 48%, 16%, and 78% of the patients received stable doses of MTX (≤ 25 mg/week), low dose corticosteroids (equivalent to ≤ 10 mg of prednisone a day), and NSAIDs, respectively. | SIMPONI 50 mg ± MTX | |
|---|---|---|
| Note: Baseline are median values. | ||
| N N reflects randomized patients; actual number of patients evaluable for each endpoint may vary by timepoint. | 113 | 146 |
| Number of swollen joints (0–66) | ||
| Baseline | 10.0 | 11.0 |
| Week 14 | 8% | 60% |
| Number of tender joints (0–68) | ||
| Baseline | 18.0 | 19.0 |
| Week 14 | 0% | 54% |
| Patient's assessment of pain (0–10) | ||
| Baseline | 5.4 | 5.8 |
| Week 14 | -1% | 48% |
| Patient's global assessment of disease activity (0–10) | ||
| Baseline | 5.2 | 5.2 |
| Week 14 | 2% | 49% |
| Physician's global assessment of disease activity (0–10) | ||
| Baseline | 5.2 | 5.4 |
| Week 14 | 7% | 59% |
| HAQ score (0–10) | ||
| Baseline | 1.0 | 1.0 |
| Week 14 | 0% | 28% |
| CRP (mg/dL) (0–10) | ||
| Baseline | 0.6 | 0.6 |
| Week 14 | 0% | 40% |
| Figure 2: Trial PsA – Percentage of ACR 20 PsA Responders by Visit: Randomized Patients The same patients may not have responded at each timepoint. |
 |
Physical Function Response in Patients with PsA
In Trial PsA, SIMPONI 50 mg demonstrated a greater improvement compared to placebo in the change in mean Health Assessment Questionnaire Disability Index (HAQ-DI) score from baseline to Week 24 (0.33 and -0.01, respectively). In addition, the SIMPONI 50 mg group compared to the placebo group had a greater proportion of HAQ responders (≥ 0.3 change from baseline) at Week 24: 43% vs. 22%, respectively.
Ankylosing Spondylitis
The safety and efficacy of SIMPONI were evaluated in a multicenter, randomized, double-blind, placebo-controlled trial in 356 adult patients with active ankylosing spondylitis according to modified New York criteria for at least 3 months (Trial AS). Patients had symptoms of active disease [defined as a Bath AS Disease Activity Index (BASDAI) ≥ 4 and VAS for total back pain of ≥ 4, on scales of 0 to 10 cm] despite current or previous NSAID therapy. Patients were excluded if they were previously treated with a biologic TNF blocker or if they had complete ankylosis of the spine. Patients were randomly assigned to placebo (N=78), SIMPONI 50 mg (N=138), or SIMPONI 100 mg (N=140) administered subcutaneously every 4 weeks. Patients were allowed to continue stable doses of concomitant MTX, sulfasalazine (SSZ), hydroxychloroquine (HCQ), low dose corticosteroids (equivalent to < 10 mg of prednisone a day), and/or NSAIDs during the trial. The use of other DMARDs including cytotoxic agents or other biologics was prohibited.
The primary endpoint was the percentage of patients achieving an Assessment in Ankylosing Spondylitis (ASAS) 20 response at Week 14. Placebo-controlled efficacy data were collected and analyzed through Week 24.
In Trial AS, the median duration of AS disease was 5.6 years, median duration of inflammatory back pain was 12 years, 83% were HLA-B27 positive, 24% had prior joint surgery or procedure, and 55% received at least one DMARD in the past. During the trial, the use of concomitant DMARDs and/or NSAIDs was as follows: MTX (20%), SSZ (26%), HCQ (1%), low dose oral steroids (16%), and NSAIDs (90%).
Clinical Response in Patients with AS
In Trial AS, SIMPONI ± DMARDs treatment, compared with placebo ± DMARDs, resulted in a significant improvement in signs and symptoms as demonstrated by the proportion of patients with an ASAS 20 response at Week 14 (see Table 7 ). There was no clear evidence of improved ASAS response with the higher SIMPONI dose group (100 mg) compared to the lower SIMPONI dose group (50 mg). Table 8 shows the percent improvement in the components of the ASAS response criteria for the SIMPONI 50 mg ± DMARDs and placebo ± DMARDs groups in Trial AS.
The percentage of patients achieving ASAS 20 responses by visit for Trial AS is shown in Figure 3. ASAS 20 responses were observed in 48% of patients in the SIMPONI 50 mg + MTX group at the first assessment (Week 4) after the initial SIMPONI administration.
| Placebo ± DMARDs During the trial, the concomitant use of stable doses of DMARDs was as follows: MTX (21%), SSZ (25%), and HCQ (1%). About 16% and 89% of patients received stable doses of low dose oral steroids and NSAIDs during the trial, respectively. | SIMPONI 50 mg ± DMARDs | |
|---|---|---|
| Bold text indicates primary endpoint. | ||
| N N reflects randomized patients. | 78 | 138 |
| Responders, % of patients | ||
| ASAS 20 | ||
| Week 14 | 22% | 59% |
| Week 24 | 23% | 56% |
| ASAS 40 | ||
| Week 14 | 15% | 45% |
| Week 24 | 15% | 44% |
| Placebo ± DMARDs During the trial, the concomitant use of stable doses of DMARDs was as follows: MTX (21%), SSZ (25%), and HCQ (1%). About 16% and 89% of patients received stable doses of low dose oral steroids and NSAIDs during the trial, respectively. | SIMPONI 50 mg ± DMARDs | |
|---|---|---|
| N N reflects randomized patients. | 78 | 138 |
| ASAS components | ||
| Patient global assessment (0–10) | ||
| Baseline | 7.2 | 7.0 |
| Week 14 | 13% | 47% |
| Total back pain (0–10) | ||
| Baseline | 7.6 | 7.5 |
| Week 14 | 9% | 50% |
| BASFI (0–10) BASFI is Bath Ankylosing Spondylitis Functional Index. | ||
| Baseline | 4.9 | 5.0 |
| Week 14 | -3% | 37% |
| Inflammation (0–10) Inflammation is the mean of 2 patient-reported stiffness self-assessments in the Bath AS Disease Activity Index (BASDAI). | ||
| Baseline | 7.1 | 7.1 |
| Week 14 | 6% | 59% |
| Figure 3: Trial AS – Percentage of AS Patients Achieving ASAS 20 Response by Visit: Randomized Patients The same patients may not have responded at each timepoint. |
 |
Adult Ulcerative Colitis
The efficacy and safety of SIMPONI was evaluated in 2 multicenter, randomized, double-blind, placebo-controlled clinical trials in adults with moderately to severely active ulcerative colitis (UC). Trial UC-1 (NCT00487539) was a 6-week induction trial. Trial UC-2 (NCT00488631) was a randomized-withdrawal maintenance trial that evaluated 456 patients who achieved clinical response with SIMPONI induction and tolerated SIMPONI treatment.
In Trial UC-1 moderately to severely active UC was, defined as a Mayo score of 6 to 12 [the Mayo score ranges from 0 to 12 and has 4 subscales that are each scored from 0 (normal) to 3 (most severe): stool frequency, rectal bleeding, findings on endoscopy, and physician global assessment]. At baseline, subjects also had an endoscopy subscore of 2 or 3 on a 3-point scale (an endoscopy score of 2 is defined by marked erythema, absent vascular pattern, friability, erosions; and a score of 3 is defined by spontaneous bleeding, ulceration). Patients were corticosteroid dependent (i.e., an inability to successfully taper corticosteroids without a return of the symptoms of UC) or had an inadequate response to or had failed to tolerate at least one of the following therapies: oral aminosalicylates, oral corticosteroids, azathioprine, or 6-mercaptopurine.
Trial UC-1 was divided into 2 parts. In Part 1 (dose finding), patients were randomized to one of 4 treatment groups: 400 mg SIMPONI administered subcutaneously (SC) at Week 0 and 200 mg at Week 2 (400/200 mg), 200 mg SIMPONI SC at Week 0 and 100 mg at Week 2 (200/100 mg), 100 mg SIMPONI SC at Week 0 and 50 mg at Week 2 (100/50 mg), or placebo SC at Weeks 0 and 2. In Part 2 (dose confirming), efficacy was evaluated in 761 patients who were randomized to receive either 400 mg SIMPONI SC at Week 0 and 200 mg at Week 2, 200 mg SIMPONI SC at Week 0 and 100 mg at Week 2, or placebo SC at Weeks 0 and 2. SIMPONI 100/50-mg SC was not evaluated in Part 2; its safety and effectiveness has not been established in UC. Concomitant stable doses of oral aminosalicylates (5-ASA), oral corticosteroids (less than 40 mg/day), azathioprine (AZA), 6-mercaptopurine (6-MP), and/or methotrexate (MTX) were permitted. Patients who received previous TNF inhibitors were excluded. The primary endpoint in UC-1 was the percent of patients in clinical response at Week 6, defined as a decrease from baseline in the Mayo score by ≥ 30% and ≥ 3 points, accompanied by a decrease in the rectal bleeding subscore of ≥ 1 or a rectal bleeding subscore of 0 (no blood seen) or 1 (streaks of blood with stool less than half the time).
Trial UC-2 evaluated 456 patients who achieved clinical response with SIMPONI induction and tolerated SIMPONI treatment. Patients were randomized to receive SIMPONI 50 mg, SIMPONI 100 mg or placebo administered subcutaneously every 4 weeks. Concomitant stable doses of oral aminosalicylates, azathioprine, 6-mercaptopurine, and/or methotrexate were permitted. Corticosteroids were to be tapered at the start of the maintenance trial. The primary endpoint in UC-2 was the percent of patients maintaining clinical response through Week 54.
Clinical Response, Clinical Remission and Improvement of Endoscopic Appearance of the Mucosa
In Trial UC-1, a greater proportion of patients achieved clinical response, clinical remission and had improvement of endoscopic appearance of the mucosa at Week 6 in the SIMPONI 200/100 mg group compared with the placebo group. The SIMPONI 400/200 mg group did not demonstrate additional clinical benefit over the SIMPONI 200/100 mg group. Clinical response was defined as a decrease from baseline in the Mayo score of ≥ 30% and ≥ 3 points, accompanied by a decrease in the rectal bleeding subscore of ≥ 1 or a rectal bleeding subscore of 0 or 1. Clinical remission was defined as a Mayo score ≤ 2 points, with no individual subscore > 1. Improvement of endoscopic appearance of the mucosa was defined as a Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern, mild friability).
In Trial UC-2, a greater proportion of patients maintained clinical response through Week 54 in the SIMPONI 100 mg group compared with the placebo group. In Trial UC-2, SIMPONI-treated patients in clinical response (which included the subset of patients in clinical remission) in Trial UC-1, were again assessed for clinical remission at Week 30 and Week 54. A greater proportion of patients had clinical remission at both Weeks 30 and 54 without demonstrating a loss of response at any time point through Week 54 in the SIMPONI 100 mg group compared with the placebo group.
These results are shown in Table 9 below.
| Trial UC-1 (6-Week Induction Trial) | |||
| Placebo N=251 | SIMPONI 200/100 mg N=253 | Treatment difference (95% CI) | |
| Clinical response Patients who had a prohibited change in concomitant UC medication, an ostomy or colectomy, discontinued trial agent due to lack of therapeutic effect, or a dose adjustment in Trial UC-2 were considered not to be in clinical response, clinical remission or have an improvement in endoscopic appearance of the mucosa from the time of the event onward. at Week 6 | 30% | 51% | 21% (12%, 29%) p<0.0001; |
| Clinical remissionat Week 6 | 6% | 18% | 11% (6%, 17%) |
| Improvement of endoscopic appearance of the mucosa at Week 6 | 29% | 42% | 14% (5%, 22%) p=0.0014; |
| Trial UC-2 (54-Week Maintenance Trial) Results in Trial UC-2 are based on patients who were in clinical response to SIMPONI at trial entry. | |||
| Placebo N=154 | SIMPONI 100 mg N=151 | Treatment difference (95% CI) | |
| Clinical responsethrough Week 54 Patients were assessed for UC disease activity by partial Mayo score every 4 weeks (loss of response was confirmed by endoscopy). Therefore, a patient who maintained clinical response was in response at each evaluation through Week 54. | 31% | 50% | 19% (8%, 29%) p<0.001; |
| Clinical remissionat both Week 30 and Week 54 A patient had to be in remission at both Weeks 30 and 54 (without demonstrating a loss of response at any time point through Week 54) to achieve sustained remission. | 16% | 28% | 12% (3%, 21%) p=0.004 |
Pediatric Ulcerative Colitis
The efficacy and safety of SIMPONI was evaluated in a multi-center, open-label pediatric study of 69 patients (NCT03596645). Efficacy was assessed in 66 patients weighing at least 15 kg with moderately to severely active ulcerative colitis defined as a Mayo score of 6 to 12 with an endoscopy subscore of ≥2 who had an inadequate response to corticosteroids, 6‑mercaptopurine (6‑MP), or azathioprine (AZA), or who were intolerant to or had medical contraindications for such therapies. Patients with prior exposure to TNF blockers were ineligible. Of the 66 subjects assessed for efficacy, the mean age was 13.4 years (range 4 to 17 years), the median weight was 51 kg, and 53% were female. Twenty-four percent identified as Hispanic or Latino; 71% identified as White, 18% as Asian, 3% as American Indian or Alaskan Native, 3% as Black, and 2% identified as part of multiple racial subgroups.
Patients weighing 15 to less than 45 kg received SIMPONI subcutaneously at 120 mg/m 2 at dosages of 120 mg/m 2 at Week 0, 60 mg/m 2 at Week 2 and 60 mg/m 2 every 4 weeks from Week 6 onward.
Patients weighing at least 45 kg received SIMPONI 200 mg at Week 0, 100 mg at Week 2 and 100 mg every 4 weeks from Week 6 onward.
The recommended body-weight tiered dosage for pediatric patients weighing 15 to less than 40 kg and 40 to less than 45 kg differs from the body surface area-based dosage administered in this study. There are no anticipated clinically relevant differences in efficacy between the recommended and studied pediatric dosages of SIMPONI [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3) ] .
At weeks 6, and 54, efficacy was assessed by the Mayo score.
Efficacy Endpoints Week 6
The results of the primary endpoint of clinical remission at Week 6 and secondary endpoint of clinical response at Week 6, both assessed by Mayo score are shown in Table 10.
| Endpoint | SIMPONI n (%) [90% CI] N=66 |
|---|---|
| Clinical remission at Week 6 Clinical remission is defined as a Mayo score ≤2 points, with no individual subscore >1. | 21 (32%) [22%, 41%] |
| Clinical response at Week 6 Clinical response is defined as a decrease from baseline in the Mayo score by ≥ 30% and ≥ 3 points, with either a decrease from baseline in the rectal bleeding subscore of ≥ 1 or a rectal bleeding subscore of 0 or 1. | 38 (58%) [48%, 68%] |
Additional Secondary Endpoints
Of the 38 pediatric subjects with clinical response at Week 6, 13 subjects (34%) were in clinical remission and 15 subjects (39%) had endoscopic improvement, defined as an endoscopy subscore of 0 or 1 based on local endoscopy, at Week 54. Of the 21 subjects in clinical remission at Week 6, 12 subjects (57%) maintained clinical remission at Week 54.
HOW SUPPLIED/STORAGE AND HANDLING
SIMPONI ® (golimumab) Injection is a preservative-free, sterile, clear to slightly opalescent, colorless to light yellow solution for subcutaneous use in a single-dose prefilled autoinjector (contains a prefilled glass syringe) or a single-dose prefilled glass syringe. The Type 1 glass syringe has a coated stopper. The fixed stainless-steel needle (5 bevel, 27G, ½ inch) is covered with a needle shield to prevent leakage of the solution through the needle and to protect the needle during handling prior to subcutaneous administration. The needle shield is made of a dry natural rubber containing latex.
| 50 mg/0.5 mL single-dose prefilled syringe | 1 pack | NDC 57894-070-01 |
| 100 mg/mL single-dose prefilled syringe | 1 pack | NDC 57894-071-01 |
| 50 mg/0.5 mL single-dose prefilled SmartJect ® autoinjector | 1 pack | NDC 57894-070-02 |
| 100 mg/mL single-dose prefilled SmartJect ® autoinjector | 1 pack | NDC 57894-071-02 |
Storage and Handling
Refrigerate SIMPONI between 36 °F to 46 °F (2 °C to 8 °C) in the original carton to protect from light until the time of use. Do not freeze. Do not shake. Do not use SIMPONI beyond the expiration date (EXP) on the carton or the expiration date on the prefilled syringe (observed through the viewing window) or the prefilled SmartJect ® autoinjector.
If needed, SIMPONI may be stored at room temperature up to 77 °F (25 °C) for a maximum single period of 30 days in the original carton to protect from light. Once a syringe or autoinjector has been stored at room temperature, do not return the product to the refrigerator. If not used within 30 days at room temperature, discard SIMPONI.
INSTRUCTIONS FOR USE
SIMPONI ® (SIM-po-nee) (golimumab) injection, for subcutaneous use
This Instructions for Use contains information on how to inject SIMPONI using the SmartJect Autoinjector.
Please read this Instructions for Use before using SIMPONI SmartJect and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your doctor about your medical condition or your treatment.
Important information
If your doctor decides that you or a caregiver may be able to give your SIMPONI injections at home, you should receive training on the right way to prepare and inject SIMPONI using SmartJect.
Do not try to inject SIMPONI yourself until you have been shown the right way to give the injections by your doctor or nurse.
Do not pinch the skin while injecting. Pinching the skin can cause failure of the device and injury, including needlestick injury.
Do not inject into the arm to avoid failure of the device or injury.
 | Store SIMPONI in the refrigerator at 36°F to 46°F (2°C to 8°C). |
Do not freeze SmartJect.
Do not shake SmartJect.
If needed, store SIMPONI at room temperature, up to 77°F (25°C) for one period of time up to 30 days. Do not return it to the refrigerator. Throw away (dispose of) if not used within 30 days at room temperature.
Keep SIMPONI in the original carton to protect from light before use.
Keep SIMPONI and all medicines out of the reach of children.
Your SmartJect at-a-glance
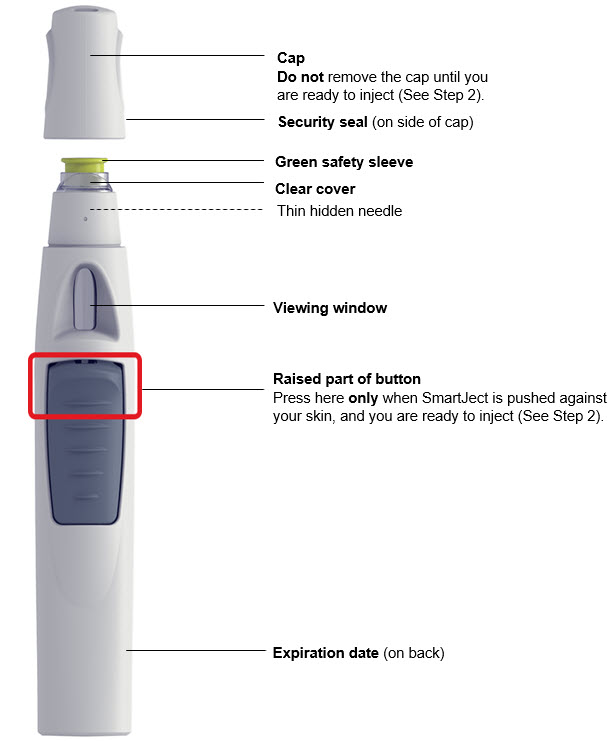
1. Preparing to inject SIMPONI using Smartject
Take out SmartJect
Take SmartJect out of the refrigerator and remove it from the carton. Place on a flat surface out of reach of children.
SmartJect should sit at room temperature for at least 30 minutes to ensure proper injection.
Do not warm any other way.
Do not remove the cap yet.
Inspect SmartJect
Check the expiration date ('EXP') on the back of SmartJect.
Do not use SIMPONI SmartJect if the expiration date has passed. Call your doctor or pharmacist for a refill.
Check the security seal on the cap.
Do not inject if the seal is broken.
Gather supplies
While SmartJect sits at room temperature for 30 minutes, gather your supplies:
- 1 Alcohol swab
- 1 Cotton ball or gauze pad
- 1 Sharps container (See Step 3 )
Check liquid in the SmartJect
After 30 minutes, check the liquid in the viewing window. It should be clear to slightly yellow and may contain tiny white or clear particles.
It is also normal to see a small air bubble.
Do not inject if the liquid is cloudy or discolored, or has large particles.
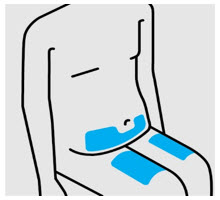
 Do not inject into the arm to avoid failure of the device or injury.
Do not inject into the arm to avoid failure of the device or injury.
Choose injection site
Select from the following areas for your injection:
- Front of thighs
- Lower abdomen ( do not use the 2-inch area around your navel (belly-button)
- Do not inject into the arms
Choose a different site within your preferred area for each injection.
Do not inject into skin that is tender, bruised, red, scaly or hard. Avoid areas with scars or stretch marks.
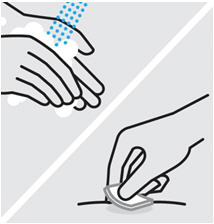
Wash hands and clean injection site
Wash your hands well with soap and warm water.
Wipe your chosen injection site with an alcohol swab and allow it to dry.
Do not touch, fan or blow on the injection site after you have cleaned it.
2. Injecting SIMPONI using SmartJect
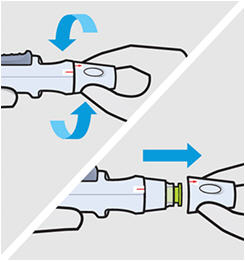
Remove cap
Twist the cap to break the security seal, then pull it straight off. Dispose of the cap right away.
It is important to inject within 5 minutes of removing the cap.
Do not put the cap back on, this may damage the hidden needle.
Do not inject if SmartJect is dropped without the cap on.
Do not pinch the skin while injecting. Pinching the skin can cause failure of the device and injury, including needlestick injury.
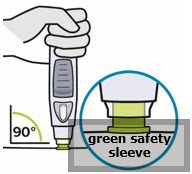
Position
Hold SmartJect comfortably with one hand above the blue button and position it straight onto your skin, as shown.
Make sure the green safety sleeve is stable and as flat as possible against your skin. If the device is not stable during the injection, you risk bending the needle.
Do not pinch your skin while positioning SmartJect onto your skin.
Do not pinch the skin to avoid injury, including needlestick injury.
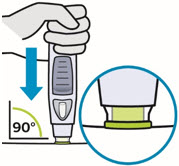
Push down
Push the open end of SmartJect against your skin at a 90-degree angle. Apply enough pressure to slide the green safety sleeve up and to keep it inside the clear cover. Only the wider part of the green safety sleeve stays outside of the clear cover.
Do not touch or press the blue button while pushing SmartJect against your skin.

The green safety sleeve helps prevent accidental injections.
Pushing the blue button before the safety sleeve is pressed down can lead to device failure.
Inject without pinching the skin.
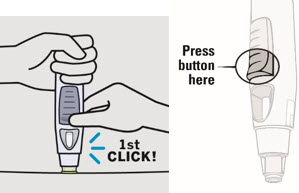
Press button and wait
Keep holding SmartJect against your skin. Use your other hand to press the raised part of the blue button to start your injection. Do not press the button until SmartJect is pressed against your skin and the safety sleeve slides into the clear cover.
You will hear a loud 1st 'click' as you press the button. This is normal, the medication is just beginning to be delivered. You may or may not feel a needle prick.
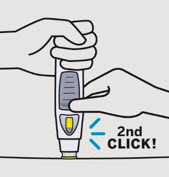
Listen for 2nd 'click'
Keep holding SmartJect against your skin until you hear the 2nd 'click' (or for 15 seconds). It usually takes about 3-6 seconds, but may take up to 15 seconds for you to hear the second 'click' sound.
The 2nd 'click' means the injection is complete and you can lift SmartJect from your skin.
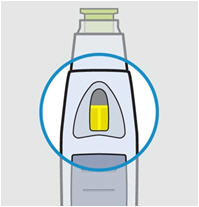
Check the viewing window
After lifting SmartJect from your skin, look for the yellow indicator in the viewing window to confirm SmartJect worked properly. The yellow indicator will fill about half of the viewing window.
If you do not see the yellow indicator, call 800-JANSSEN (800-526-7736).
Do not administer a second dose without speaking to your doctor.
3. Disposing of SIMPONI SmartJect
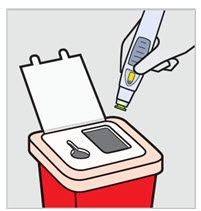
Dispose of your SmartJect
Put your used SmartJect in an FDA-cleared sharps disposal container right away after use.
Do not recycle your used sharps disposal container.
For more information, see " Helpful tips ".
Check injection site
There may be a small amount of blood or liquid at the injection site.
Hold pressure over your skin with a cotton ball or gauze pad until any bleeding stops.
Do not rub the injection site.
If needed, cover injection site with a bandage. Your injection is now complete!
 Need help?
Need help?
Call your doctor or pharmacist to talk about any questions you may have. For additional assistance or to share your feedback call 800-JANSSEN (800-526-7736).
Helpful tips If you are having difficulty injecting:
|
Additional Disposal Information If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: www.fda.gov/safesharpsdisposal |
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by: Janssen Biotech, Inc. Horsham, PA 19044, USA US License No. 1864
Revised: September 2025
Mechanism of Action
Golimumab is a human monoclonal antibody that binds to both the soluble and transmembrane bioactive forms of human TNFα. This interaction prevents the binding of TNFα to its receptors, thereby inhibiting the biological activity of TNFα (a cytokine protein). There was no evidence of the golimumab antibody binding to other TNF superfamily ligands; in particular, the golimumab antibody did not bind or neutralize human lymphotoxin. Golimumab did not lyse human monocytes expressing transmembrane TNF in the presence of complement or effector cells.
Elevated TNFα levels in the blood, synovium, and joints have been implicated in the pathophysiology of several chronic inflammatory diseases such as rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. TNFα is an important mediator of the articular inflammation that is characteristic of these diseases. The exact mechanism by which golimumab treats ulcerative colitis is unknown. Golimumab modulated the in vitro biological effects mediated by TNF in several bioassays, including the expression of adhesion proteins responsible for leukocyte infiltration (E-selectin, ICAM-1 and VCAM-1) and the secretion of proinflammatory cytokines (IL-6, IL-8, G-CSF and GM-CSF).