Get your patient on Skytrofa (Lonapegsomatropin-Tcgd)
Skytrofa patient education
Patient toolkit
Dosage & administration

Skytrofa prescribing information
INDICATIONS AND USAGE
SKYTROFA (lonapegsomatropin-tcgd) is a human growth hormone indicated for the:
- Treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
- Replacement of endogenous growth hormone in adults with growth hormone deficiency (GHD).
DOSAGE AND ADMINISTRATION
- SKYTROFA should be administered subcutaneously once weekly into the abdomen, buttock, or thigh with regular rotation of the injection sites (2.7 ).
- Pediatric Patients: Recommended dose is 0.24 mg/kg body weight once weekly (2.2 ).
- Adults: Recommended starting dose is based on age and concomitant use of oral estrogen. Titrate monthly until the desired clinical response and/or weekly average IGF-1 concentration are achieved (2.3 ).
- See Full Prescribing Information for instructions on preparation and administration of drug (2.5 , 2.6 , 2.7 ).
General Dosing Information
- For subcutaneous injection, once weekly.
- Therapy with SKYTROFA should be supervised by a healthcare provider who is experienced in the diagnosis and management of patients with growth hormone deficiency (GHD).
- Perform fundoscopic examination before initiating treatment with SKYTROFA to exclude preexisting papilledema. If papilledema is identified, evaluate the etiology and treat the underlying cause before initiating treatment with SKYTROFA [see Warnings and Precautions (5.5) ] .
Recommended Dosage for Pediatric Patients
- The recommended dose of SKYTROFA for treatment-naïve patients and patients switching from another growth hormone product is 0.24 mg/kg body weight, given once weekly.
- Individualize and titrate the dosage of SKYTROFA based on response.
- When changing from daily somatropin therapy to once-weekly SKYTROFA, wait at least 8 hours between the final dose of daily somatropin and the first dose of SKYTROFA.
- When changing from another once-weekly growth hormone therapy to once-weekly SKYTROFA, wait at least 7 days between the final dose of the previous growth hormone therapy and the first dose of SKYTROFA.
- Assess compliance and evaluate other causes of poor growth, such as hypothyroidism, under-nutrition, advanced bone age and antibodies to recombinant human growth hormone if patients experience failure to increase height velocity, particularly during the first year of treatment.
- Patients who were treated with SKYTROFA for GH deficiency in childhood and whose epiphyses are closed should be reevaluated before continuing SKYTROFA.
Recommended Dosage for Adults
- The recommended starting dose of SKYTROFA in adults with GHD is based on age and concomitant use of oral estrogen [see Dosage Forms and Strengths (3) ] . For treatment-naïve patients or for patients switching from another growth hormone product, start SKYTROFA as described below:
- 1.4 mg once weekly for adults 30 to 60 years old, with no oral estrogen intake
- 2.1 mg once weekly for adults under 30 years old, or adults of any age intaking oral estrogen
- 0.7 mg once weekly for adults over 60 years old, with no oral estrogen intake
- When changing from daily somatropin therapy to once-weekly SKYTROFA, wait at least 8 hours between the final dose of daily somatropin and the first dose of SKYTROFA.
- When changing from another once-weekly growth hormone therapy to once-weekly SKYTROFA, wait at least 7 days between the final dose of the previous growth hormone therapy and the first dose of SKYTROFA.
- Increase the dose monthly to a higher strength cartridge based on the clinical response and/or IGF-1 concentration. Draw IGF-1 serum sample 4 to 5 days after the prior dose.
- Decrease the dose to a lower strength cartridge as needed based on adverse reactions or a weekly average IGF-1 concentration above the age- and sex-specific normal range.
- The maximum recommended dose is 6.3 mg once weekly.
Missed Doses
- Administer a missed dose as soon as possible and not more than 2 days after the missed dose.
- To avoid missed doses, SKYTROFA can be taken 2 days before or 2 days after the scheduled dosing day. Resume once-weekly dosing for the next dose at the previously scheduled dosing day.
- If more than 2 days have passed from the scheduled day, skip the dose and administer the next dose on the regularly scheduled day.
- At least 5 days should elapse between doses.
Administration Instructions for Pediatric Patients
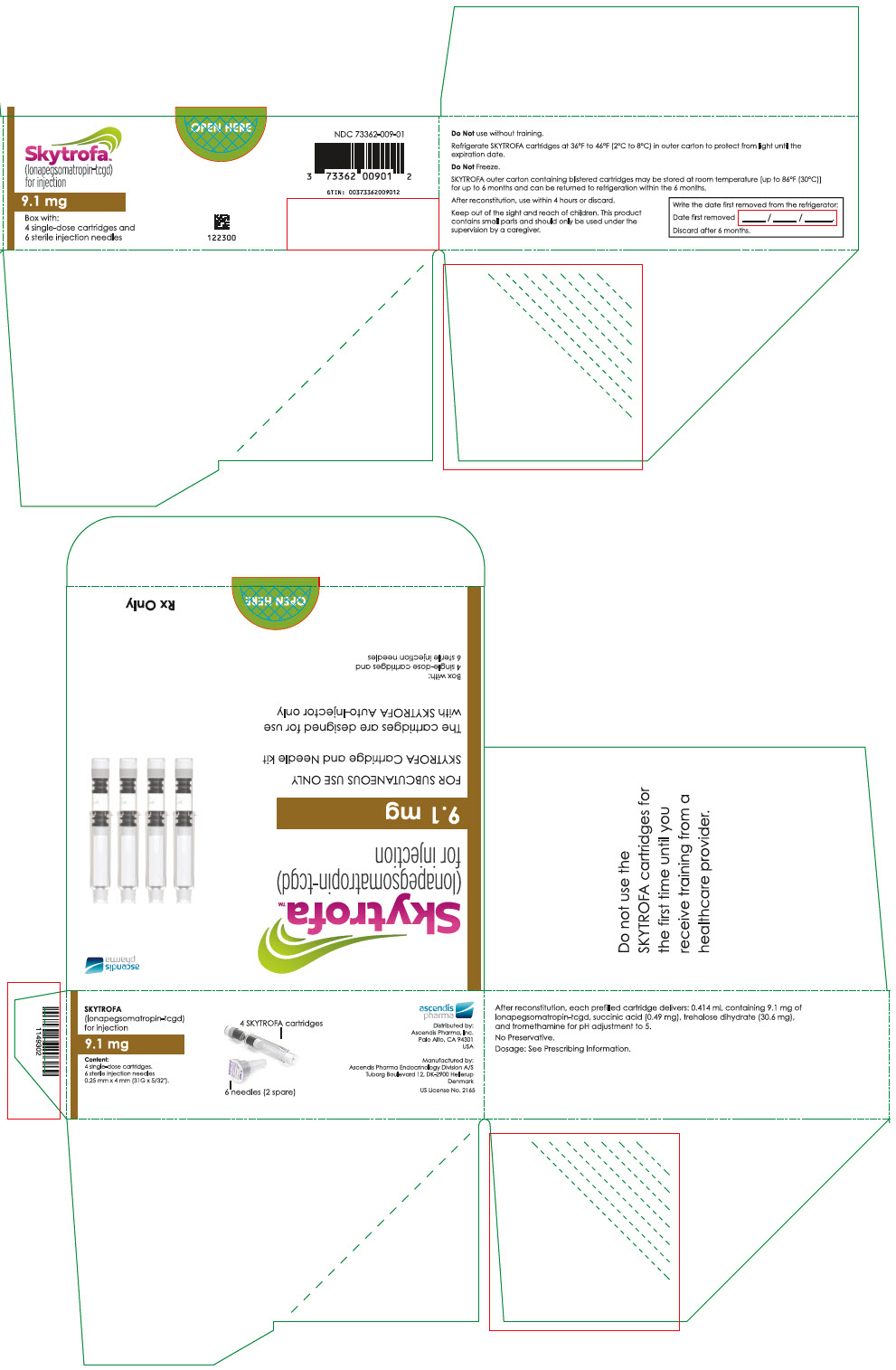
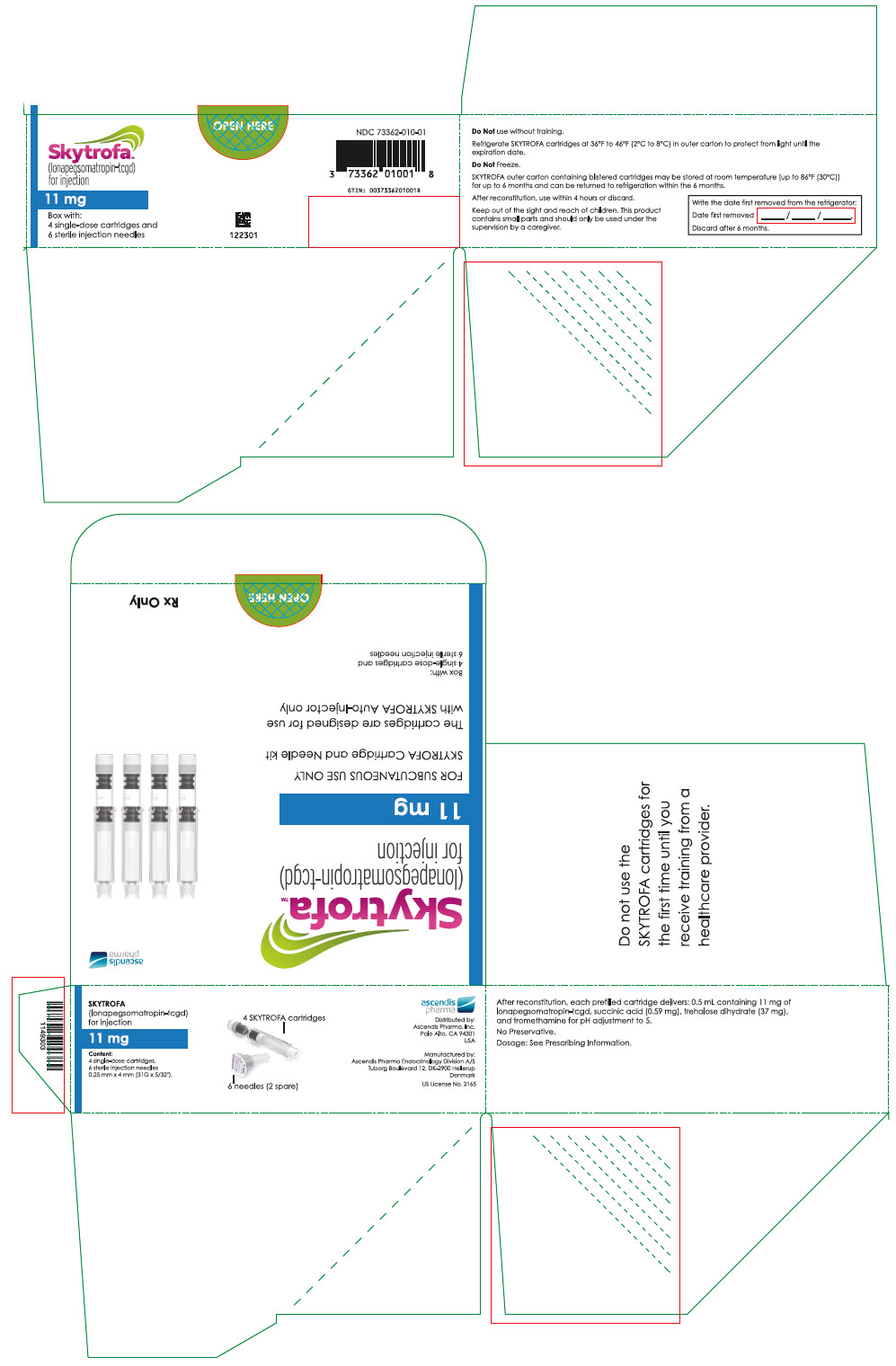
SKYTROFA is available in 9 cartridges (dosage strengths in somatropin equivalents) for pediatric patients.
Selection of the appropriate cartridge (mg) is based on the prescribed dose (mg/kg) and the patient's body weight (kg) [see Dosage Forms and Strengths (3) ] .
- If prescribing a dose of 0.24 mg/kg/week and the patient's weight is 11.5 to 100 kg, follow the recommended dosing in Table 1.
- If prescribing a dose other than 0.24 mg/kg/week, calculate the total weekly dose (in mg) and select the appropriate cartridge as follows:
- Total weekly dose (mg) = prescribed weekly dose (mg/kg) × patient's body weight (kg).
- Round the total weekly dose (mg) to the closest cartridge dose while also considering treatment goals and clinical response.
| Weight (kg) | Dose (mg) |
|---|---|
| 11.5 – 13.9 | 3 |
| 14 – 16.4 | 3.6 |
| 16.5 – 19.9 | 4.3 |
| 20 – 23.9 | 5.2 |
| 24 – 28.9 | 6.3 |
| 29 – 34.9 | 7.6 |
| 35 – 41.9 | 9.1 |
| 42 – 50.9 | 11 |
| 51 – 60.4 | 13.3 |
| 60.5 – 69.9 | 15.2 (using two cartridges of 7.6 mg each) |
| 70 – 84.9 | 18.2 (using two cartridges of 9.1 mg each) |
| 85 – 100 | 22 (using two cartridges of 11 mg each) |
Administration Instructions for Adults
SKYTROFA is available in 14 cartridges (dosage strengths in somatropin equivalents) for adults.
Selection of the appropriate cartridge (mg) is based on the prescribed dose (mg/week) [see Dosage Forms and Strengths (3) ] .
Preparation and Administration
- The SKYTROFA cartridge has been designed for use only with the SKYTROFA Auto-Injector.
- If refrigerated, the SKYTROFA cartridge must be kept at room temperature for 15 minutes before use.
- The SKYTROFA Auto-Injector provides a fully automated reconstitution of the lyophilized drug product which is followed by a manual mixing step controlled by the device. When the injection needle is inserted into the skin, the device automatically delivers the drug product. The built-in electronics and software assist the user during the entire preparation and injection sequence and provide confirmation that the full dose has been delivered.
- The mixed solution should be clear and colorless to opalescent. The solution may contain air bubbles and this is acceptable. DO NOT inject if the solution is cloudy or contains particulate matter.
- Use SKYTROFA cartridges within 4 hours after reconstitution. Discard reconstituted SKYTROFA cartridges after 4 hours when stored at room temperature up to 86°F (30°C).
- Inject SKYTROFA subcutaneously into the abdomen, buttock, or thigh. Rotate injection sites between and within regions to reduce the risk of lipoatrophy [see Warnings and Precautions (5.12) ] .
- Refer to the Instructions for Use for complete administration instructions with illustrations. The instructions can also be found on www.Skytrofa.com/IFU.
- Patients and/or caregivers who will administer SKYTROFA should receive appropriate training and instruction on the proper use of SKYTROFA from their healthcare provider.
DOSAGE FORMS AND STRENGTHS
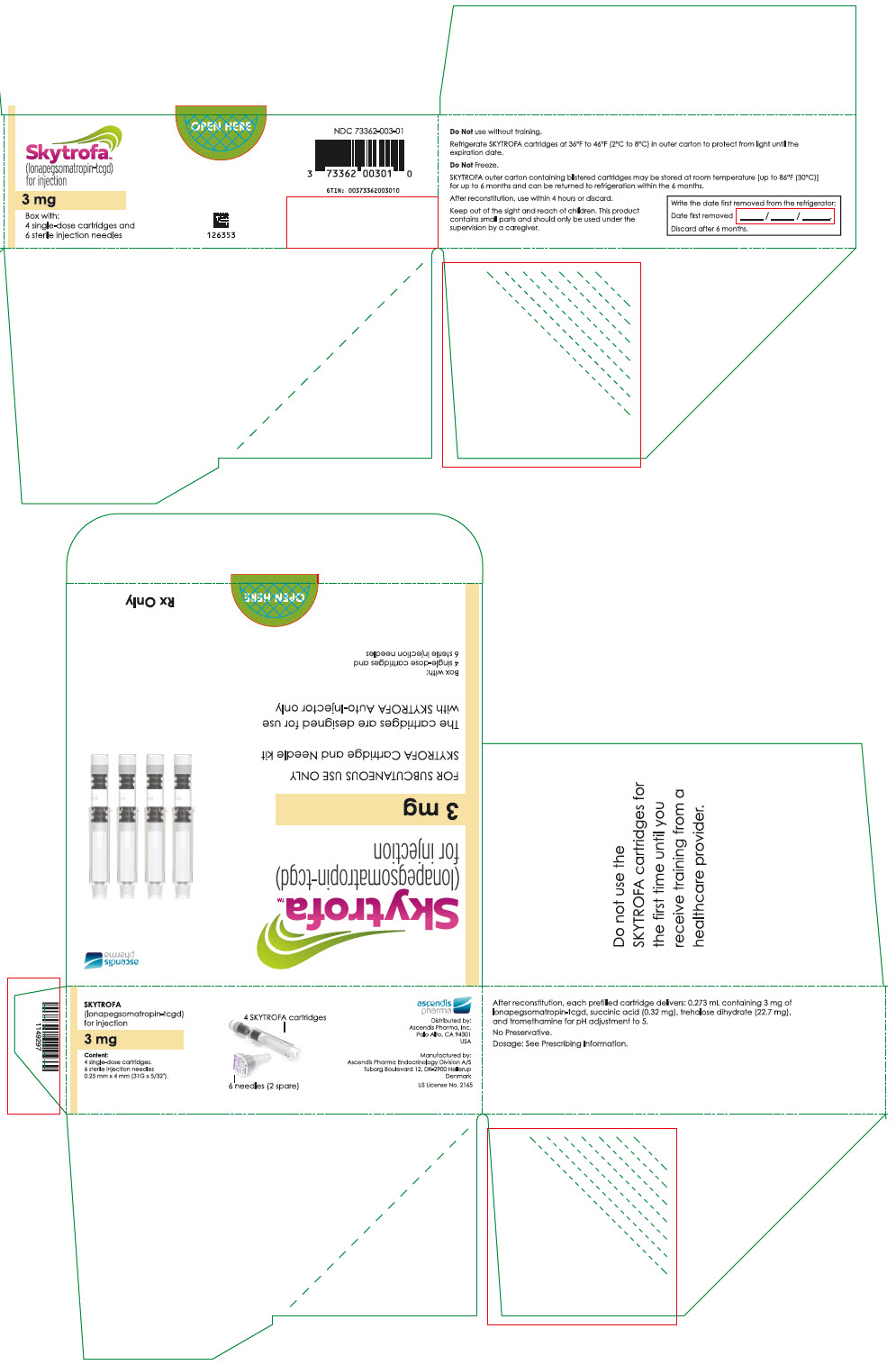
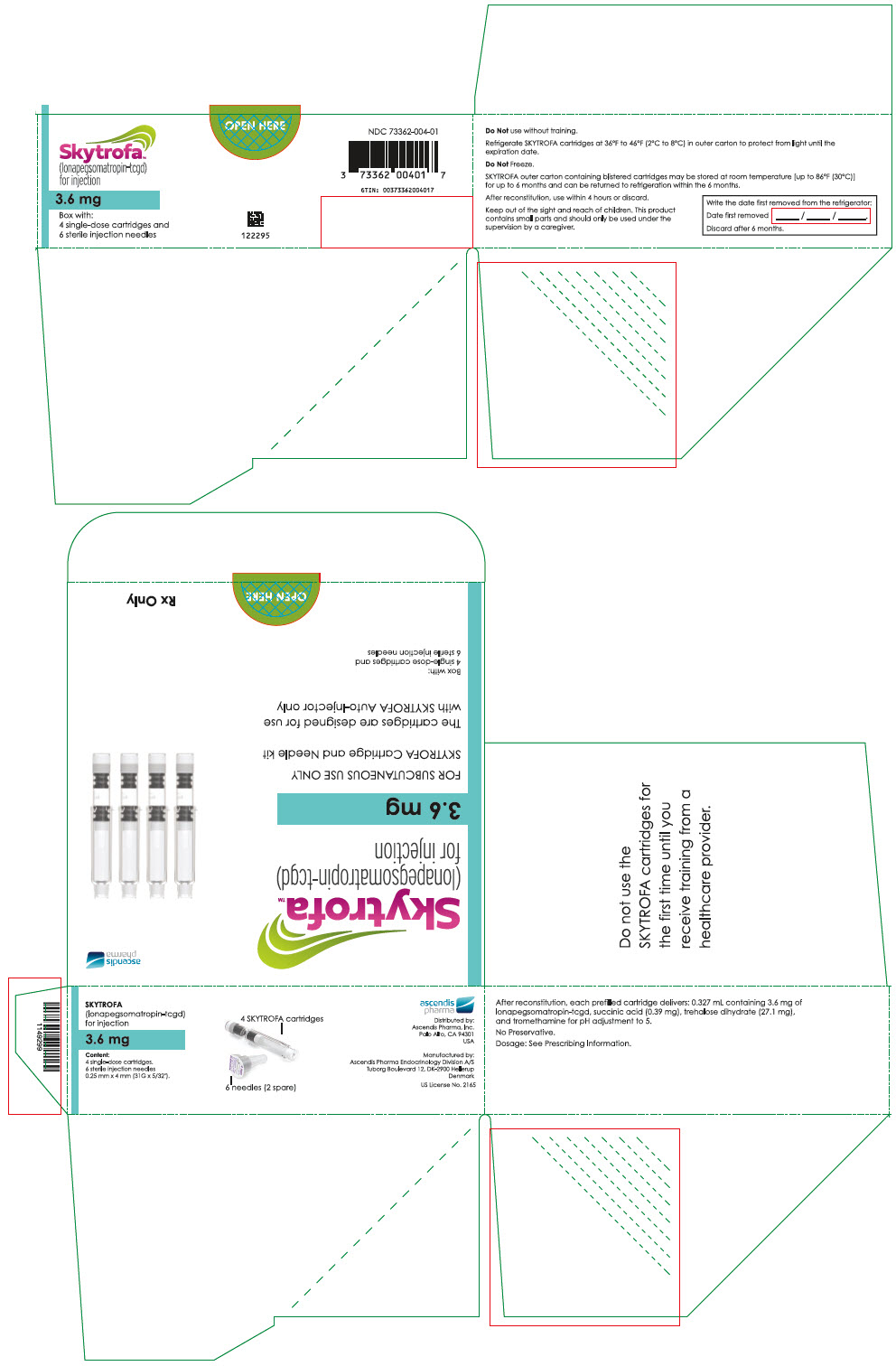
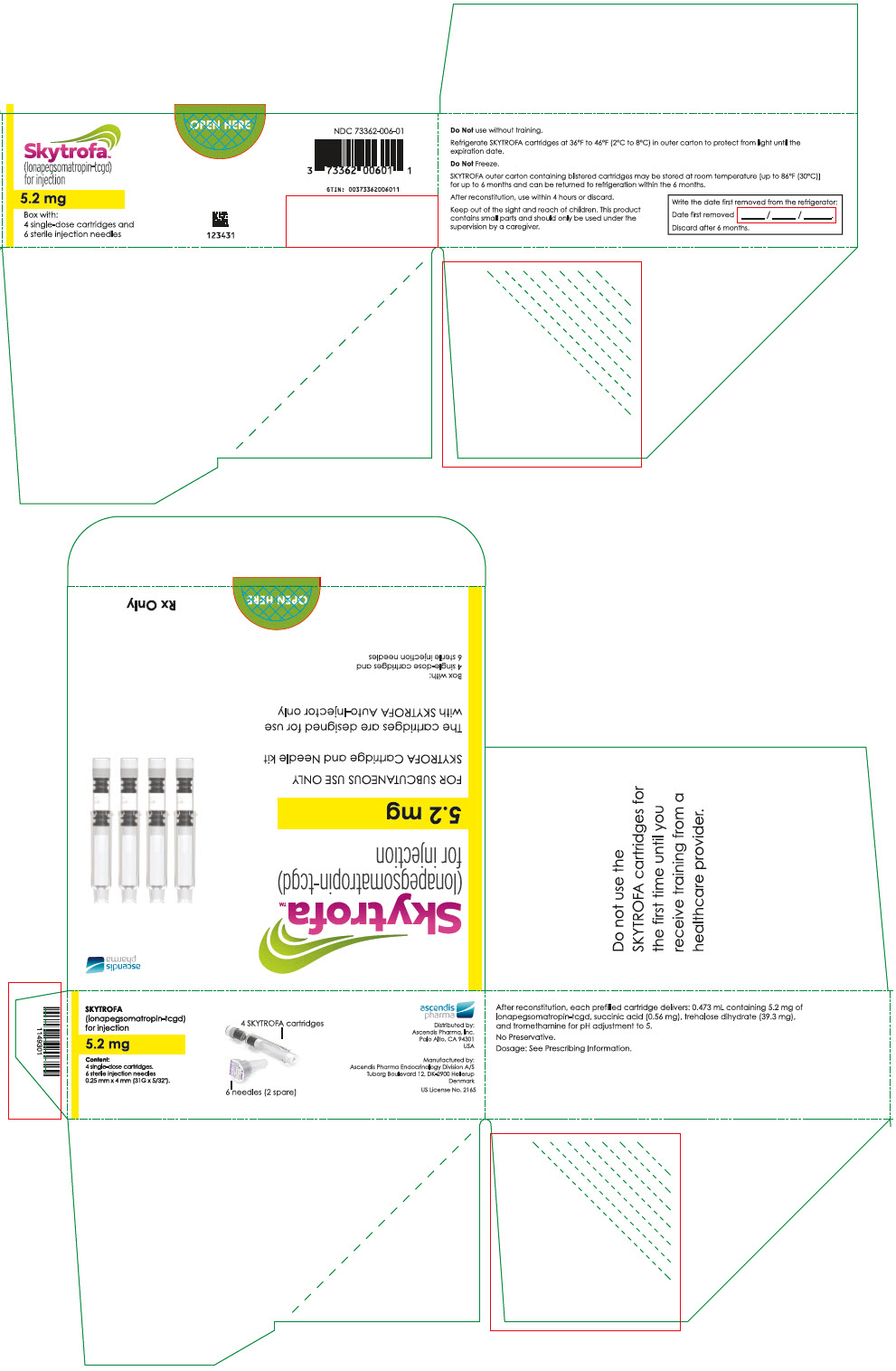
SKYTROFA is a white to off-white lyophilized powder available in a single-dose, dual-chamber, prefilled cartridge containing lonapegsomatropin-tcgd in one chamber and diluent, Water for Injection, in the other chamber and is available in the following strengths:
For injection: 0.7 mg, 1.4 mg, 1.8 mg, 2.1 mg, 2.5 mg, 3 mg, 3.6 mg, 4.3 mg, 5.2 mg, 6.3 mg, 7.6 mg, 9.1 mg, 11 mg, and 13.3 mg.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
There are no available data on lonapegsomatropin-tcgd use in pregnant patients to evaluate a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Available published data over several decades for somatropin, the active component of lonapegsomatropin-tcgd, have not identified a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, there was no evidence of embryo-fetal or neonatal harm when pregnant rats were administered subcutaneous lonapegsomatropin-tcgd at doses up to 13-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 30-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week (see Data ) .
The estimated background risk of birth defects and miscarriages for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
No embryonic or fetal development toxicities occurred in rats administered subcutaneous lonapegsomatropin-tcgd at doses up to 13-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 30-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week.
In a peri- and post-natal developmental study in rats, there were no adverse effects on the pregnant/lactating female or on development of the conceptus and the offspring following exposure of the female from implantation through weaning to doses of a structurally related pegylated somatropin prodrug up to 13-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 30-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week.
Lactation
Risk Summary
There are no data on the presence of lonapegsomatropin-tcgd in human milk, effects on the breastfed infant, or effects on milk production. High molecular weight therapeutic proteins, including lonapegsomatropin-tcgd, are expected to have low passage into human milk and limited systemic exposure in the breastfed infant. Additionally, published data indicate that exogenous somatropin does not increase normal human milk concentrations of growth hormone. No adverse effects on the breastfed infant have been reported with somatropin. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SKYTROFA and any potential adverse effects on the breastfed infant from SKYTROFA or from the underlying maternal condition.
Pediatric Use
Safety and effectiveness of SKYTROFA have been established in pediatric patients 1 year and older and who weigh at least 11.5 kg. Pediatric use was established in a controlled study of 161 treatment-naïve pediatric patients ages 3 to 13 years and by supportive data in pediatric patients 1 year and older [see Adverse Reactions (6) and Clinical Studies (14) ] .
The safety and effectiveness of SKYTROFA in children less than 1 year of age have not been established.
Use of somatropin in pediatric patients with Prader-Willi syndrome has been associated with reports of sudden death. SKYTROFA is not indicated for the treatment of pediatric patients with growth failure due to genetically confirmed Prader-Willi syndrome [see Warnings and Precautions (5.13) ] .
Geriatric Use
Of the 249 patients who received SKYTROFA in clinical studies, 24 (10%) patients were 65 years of age and older, and 4 (2%) patients were 75 years of age and older. Geriatric patients may be at an increased risk for adverse reactions. Initiate SKYTROFA at 0.7 mg once weekly in patients 60 years of age and older, not on estrogen therapy [see Dosage and Administration (2.3) ] .
CONTRAINDICATIONS
SKYTROFA is contraindicated in patients with:
- Acute critical illness after open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure due to the risk of increased mortality with use of pharmacologic doses of somatropin [see Warnings and Precautions (5.1) ] .
- Hypersensitivity to somatropin or any of the excipients in SKYTROFA. Severe systemic hypersensitivity reactions, including anaphylactic reactions and angioedema, have been reported [see Warnings and Precautions (5.2) ].
- Pediatric patients with closed epiphyses.
- Active malignancy due to the risk of malignancy progression [see Warnings and Precautions (5.3) ].
- Active proliferative or severe non-proliferative diabetic retinopathy because treatment with somatropin may worsen this condition.
- Pediatric patients with Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea or have severe respiratory impairment due to the risk of sudden death [see Warnings and Precautions (5.13) ].
WARNINGS AND PRECAUTIONS
- Severe Hypersensitivity: Hypersensitivity reactions, including anaphylaxis and angioedema have occurred. In the event of an allergic reaction, seek prompt medical attention (5.2 ).
- Increased Risk of Neoplasms: Monitor patients with preexisting tumors for progressions or recurrence. Increased risk of a second neoplasm in childhood cancer survivors treated with somatropin – in particular meningiomas in patients treated with radiation to the head for their first neoplasm (5.3 ).
- Glucose Intolerance and Diabetes Mellitus: May be unmasked. Periodically monitor glucose levels in all patients. Doses of concurrent antihyperglycemic drugs in diabetics may require adjustment (5.4 ).
- Intracranial Hypertension: Exclude preexisting papilledema. May develop and is usually reversible after discontinuation or dose reduction (5.5 ).
- Fluid Retention (i.e., edema, arthralgia, carpal tunnel syndrome): May occur. Reduce dose as necessary (5.6 ).
- Hypoadrenalism: Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism (5.7 ).
- Hypothyroidism: May first become evident or worsen (5.8 ).
- Slipped Capital Femoral Epiphysis in Pediatric Patients: May develop. Evaluate children with the onset of a limp or persistent hip/knee pain (5.9 ).
- Progression of Preexisting Scoliosis: May develop (5.10 ).
- Pancreatitis: Consider pancreatitis in patients with persistent severe abdominal pain (5.11 ).
Increased Mortality in Patients With Acute Critical Illness
Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin [see Contraindications (4) ].
The safety of continuing SKYTROFA treatment in patients receiving replacement doses for the approved indication who concurrently develop these illnesses has not been established.
Severe Hypersensitivity
Severe systemic hypersensitivity reactions, including anaphylactic reactions and angioedema have been reported with postmarketing use of somatropin products, including SKYTROFA. Inform patients and/or caregivers that such reactions are possible, and that prompt medical attention should be sought if an allergic reaction occurs. SKYTROFA is contraindicated in patients with known hypersensitivity to somatropin or any of the excipients in SKYTROFA.
Increased Risk of Neoplasms
Active Malignancy
There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy [see Contraindications (4) ]. Any preexisting malignancy should be inactive, and its treatment should be completed prior to instituting therapy with SKYTROFA. Discontinue SKYTROFA if there is evidence of recurrent malignancy.
Risk of Second Neoplasm in Pediatric Patients
In childhood cancer survivors who were treated with radiation to the brain/head for their first neoplasm and who developed subsequent growth hormone deficiency (GHD) and were treated with somatropin, an increased risk of a second neoplasm has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. Monitor all patients with a history of GHD secondary to an intracranial neoplasm while on somatropin therapy for progression or recurrence of the tumor.
New Malignancy During Treatment
Because children with certain rare genetic causes of short stature have an increased risk of developing malignancies, thoroughly consider the risks and benefits of starting somatropin in these patients. If treatment with somatropin is initiated, carefully monitor these patients for development of neoplasms.
Monitor patients on SKYTROFA therapy carefully for increased growth or potential malignant changes of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of preexisting nevi.
Glucose Intolerance and Diabetes Mellitus
Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. Previously undiagnosed impaired glucose tolerance and overt type 2 diabetes mellitus may be unmasked. Monitor glucose levels in all patients receiving SKYTROFA, especially in those with risk factors for type 2 diabetes mellitus, such as obesity or a family history of type 2 diabetes mellitus. When initiating SKYTROFA, monitor closely patients with preexisting type 1 or type 2 diabetes mellitus or impaired glucose tolerance and adjust the doses of antihyperglycemic drugs as needed.
Intracranial Hypertension
Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropin. Symptoms usually occurred within 8 weeks after the initiation of somatropin. In all reported cases, IH-associated signs and symptoms resolved rapidly after cessation of therapy or a reduction of the somatropin dose. To exclude preexisting papilledema, perform fundoscopic examination before initiating treatment with SKYTROFA, and reassess periodically thereafter. If papilledema is observed by fundoscopy, stop somatropin treatment. If somatropin-induced IH is confirmed, restart treatment with SKYTROFA at a lower dose after IH-associated signs and symptoms have resolved.
Fluid Retention
Fluid retention during somatropin therapy may occur. Clinical manifestations of fluid retention (e.g., edema, arthralgia, myalgia, nerve compression syndromes, including carpal tunnel syndrome/paresthesia) are usually transient and dose dependent.
Hypoadrenalism
Patients receiving somatropin therapy who have or are at risk for pituitary hormone deficiency(s) may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. In addition, patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA therapy. Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in patients with known hypoadrenalism [see Drug Interactions (7) ] .
Hypothyroidism
Undiagnosed or untreated hypothyroidism may prevent optimal response to SKYTROFA. In patients with GHD, central (secondary) hypothyroidism may first become evident or worsen during SKYTROFA treatment. Therefore, perform periodic thyroid function tests in patients and initiate or appropriately adjust thyroid hormone replacement therapy when indicated.
Slipped Capital Femoral Epiphysis in Pediatric Patients
Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Slipped capital femoral epiphysis may lead to osteonecrosis. Cases of slipped capital femoral epiphysis with or without osteonecrosis have been reported in pediatric patients with short stature receiving somatropin. Evaluate pediatric patients receiving SKYTROFA with the onset of a limp or complaints of persistent hip or knee pain for slipped capital femoral epiphysis and osteonecrosis and manage accordingly.
Progression of Preexisting Scoliosis in Pediatric Patients
Somatropin increases growth rate in pediatric patients, and progression of existing scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for disease progression.
Pancreatitis
Pancreatitis has been reported in pediatric patients receiving somatropin. The risk may be greater in pediatric patients than adults. Consider pancreatitis in patients who develop persistent severe abdominal pain.
Lipoatrophy
When SKYTROFA is administered subcutaneously at the same site over a long period of time, lipoatrophy may result. Rotate injection sites when administering SKYTROFA to reduce this risk [see Dosage and Administration (2.7) ] .
Sudden Death in Pediatric Patients With Prader-Willi Syndrome
There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. SKYTROFA is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
Laboratory Tests
Serum levels of alkaline phosphatase and phosphate may increase after SKYTROFA therapy. Serum levels of parathyroid hormone may increase after somatropin treatment. If a patient is found to have abnormal laboratory tests, monitor as appropriate.
ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in the labeling:
- Increased mortality in patients with acute critical illness [see Warnings and Precautions (5.1) ]
- Severe hypersensitivity [see Warnings and Precautions (5.2) ]
- Increased risk of neoplasms [see Warnings and Precautions (5.3) ]
- Glucose intolerance and diabetes mellitus [see Warnings and Precautions (5.4) ]
- Intracranial hypertension [see Warnings and Precautions (5.5) ]
- Fluid retention [see Warnings and Precautions (5.6) ]
- Hypoadrenalism [see Warnings and Precautions (5.7) ]
- Hypothyroidism [see Warnings and Precautions (5.8) ]
- Slipped capital femoral epiphysis in pediatric patients [see Warnings and Precautions (5.9) ]
- Progression of preexisting scoliosis in pediatric patients [see Warnings and Precautions (5.10) ]
- Pancreatitis [see Warnings and Precautions (5.11) ]
- Lipoatrophy [see Warnings and Precautions (5.12) ]
- Sudden death in pediatric patients with Prader-Willi syndrome [see Warnings and Precautions (5.13) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Pediatric Patients with Growth Hormone Deficiency
SKYTROFA was studied in a 52-week, open-label, active-controlled trial in 161 treatment-naïve, prepubertal pediatric patients with growth hormone deficiency (GHD) [see Clinical Studies (14.1) ] . The subjects ranged in age from 3.2 to 13.1 years with a mean of 8.5 years. One hundred thirty-two (82%) of the subjects were male and 29 (18%) were female. One subject was Asian, 3 were Black or African American, 152 were Caucasian, and 5 were categorized as "other."
Table 2 shows common adverse reactions that occurred in ≥ 5% of patients treated with SKYTROFA in this trial.
| Adverse reactions | Daily Somatropin (N = 56) n (%) | SKYTROFA (N = 105) n (%) |
|---|---|---|
| Adverse reactions that are medically related were grouped to a single preferred term. | ||
| Infection, viral | 6 (11%) | 16 (15%) |
| Pyrexia | 5 (9%) | 16 (15%) |
| Cough | 4 (7%) | 11 (11%) |
| Nausea and vomiting | 4 (7%) | 11 (11%) |
| Hemorrhage Hemorrhage in the SKYTROFA treatment group included epistaxis (3), contusion (2), petechiae (1) and eye hemorrhage (1). | 1 (2%) | 7 (7%) |
| Diarrhea | 3 (5%) | 6 (6%) |
| Abdominal pain | 2 (4%) | 6 (6%) |
| Arthralgia and arthritis Arthralgia and arthritis in the SKYTROFA treatment group included arthralgia (5) and reactive arthritis (1). | 1 (2%) | 6 (6%) |
Laboratory Tests
More SKYTROFA-treated patients shifted from normal baseline levels to elevated phosphate and alkaline phosphatase levels at the end of the trial compared to the daily somatropin group (44.2% vs. 30.2% and 19.2% vs. 9.4%, respectively); these laboratory changes occurred intermittently [see Warnings and Precautions (5.14) ] .
Adults with Growth Hormone Deficiency
SKYTROFA was studied in a 38-week parallel-arm, placebo-controlled (double-blind) and active-controlled (open label) trial in 259 adults with growth hormone deficiency (GHD) [see Clinical Studies (14.2) ] .
The mean (range) age at enrollment was 43 (23 to 81) years old, with 119 (46%) females (55 on oral estrogen) and 140 (54%) males. One subject was American Indian or Alaska Native, one was Black or African American, 28 were Asian, and 218 were Caucasian.
Table 3 shows adverse reactions that occurred in ≥ 5% of adults treated with SKYTROFA and more frequently than in placebo-treated adults in this trial.
| Adverse reactions | Placebo (N = 84) n (%) | SKYTROFA (N = 89) n (%) |
|---|---|---|
| Adverse reactions that are medically related were grouped to a single preferred term. | ||
| Edema Edema in the SKYTROFA treatment group included edema peripheral (6) and peripheral swelling (1). | 1 (1%) | 7 (8%) |
| Central (secondary) hypothyroidism Central (secondary) hypothyroidism in the SKYTROFA treatment group included thyroxine free decreased (3), central hypothyroidism (2), thyroxine decreased (1), blood thyroid stimulating hormone decreased (1), tri-iodothyronine free decreased (1). Preexisting central hypothyroidism in 5 of 6 SKYTROFA-treated patients. | 1 (1%) | 6 (7%) |
Laboratory Tests
More SKYTROFA-treated patients shifted from normal or low baseline levels to elevated alkaline phosphatase levels at the end of the trial compared to the placebo group (14% vs. 6%); these laboratory changes were noted with increased frequency as the trial progressed [see Warnings and Precautions (5.14) ] .
Postmarketing Experience
The following adverse reactions have been identified during post approval use of somatropin products or SKYTROFA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Severe systemic hypersensitivity reactions, including anaphylactic reactions and angioedema
- Musculoskeletal and connective tissue disorders – osteonecrosis in pediatric patients
DRUG INTERACTIONS
Table 4 includes a list of drugs with clinically important drug interactions when administered concomitantly with SKYTROFA and instructions for preventing or managing them.
| Replacement Glucocorticoid Treatment | |
| Clinical Impact: | Microsomal enzyme 11β-hydroxysteroid dehydrogenase type 1 (11βHSD-1) is required for conversion of cortisone to its active metabolite, cortisol, in hepatic and adipose tissue. Somatropin inhibits 11βHSD-1. Consequently, individuals with untreated growth hormone deficiency (GHD) have relative increases in 11βHSD-1 and serum cortisol. Initiation of SKYTROFA may result in inhibition of 11βHSD-1 and reduced serum cortisol concentrations. |
| Intervention: | Patients treated with glucocorticoid replacement for hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA [see Warnings and Precautions (5.7) ] |
| Examples | Cortisone acetate and prednisone may be affected more than others because conversion of these drugs to their biologically active metabolites is dependent on the activity of 11βHSD-1. |
| Pharmacologic Glucocorticoid Therapy and Supraphysiologic Glucocorticoid Treatment | |
| Clinical Impact: | Pharmacologic glucocorticoid therapy and supraphysiologic glucocorticoid treatment may attenuate the growth-promoting effects of SKYTROFA in pediatric patients. |
| Intervention: | Carefully adjust glucocorticoid replacement dosing in pediatric patients receiving glucocorticoid treatments to avoid both hypoadrenalism and an inhibitory effect on growth. |
| Cytochrome P450-Metabolized Drugs | |
| Clinical Impact: | Limited published data indicate that somatropin treatment increases cytochrome P450 (CYP450)-mediated antipyrine clearance. SKYTROFA may alter the clearance of compounds known to be metabolized by CYP450 liver enzymes. |
| Intervention: | Careful monitoring is advisable when SKYTROFA is administered in combination with drugs metabolized by CYP450 liver enzymes. |
| Oral Estrogen | |
| Clinical Impact: | Oral estrogens may reduce the serum insulin-like growth factor-1 (IGF-1) response to SKYTROFA. |
| Intervention: | Patients receiving oral estrogen replacement may require higher SKYTROFA dosages. |
| Insulin and/or Other Antihyperglycemic Agents | |
| Clinical Impact: | Treatment with SKYTROFA may decrease insulin sensitivity, particularly at higher doses. |
| Intervention: | Patients with diabetes mellitus may require adjustment of their doses of insulin and/or other antihyperglycemic agents [see Warnings and Precautions (5.4) ] . |
DESCRIPTION
Lonapegsomatropin-tcgd is a long-acting prodrug of a human growth hormone (somatropin) produced by recombinant DNA technology using E. coli . Lonapegsomatropin-tcgd consists of a parent drug, somatropin, that is conjugated to a methoxypolyethylene glycol carrier (4 × 10 kDa mPEG) via a proprietary TransCon Linker and has a molecular weight of 63 kDa (released somatropin is 22 kDa). In vitro assay confirms the minimum potency of released somatropin is NLT 2.5 IU/mg.
SKYTROFA (lonapegsomatropin-tcgd) for injection is a sterile, preservative-free, white to off-white lyophilized powder available in a single-dose, dual-chamber, prefilled cartridge containing lonapegsomatropin-tcgd in one chamber and the diluent, Water for Injection, in the other chamber. SKYTROFA prefilled cartridge must be used with SKYTROFA Auto-Injector to provide an automatic mixing step for reconstitution prior to subcutaneous use.
After reconstitution, each prefilled cartridge delivers:
- 0.327 mL containing 0.7 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg), trehalose dihydrate (29.8 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 1.4 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (29.1 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 1.8 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (28.8 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 2.1 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (28.4 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 2.5 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg) trehalose dihydrate (28.1 mg) and tromethamine for pH adjustment to 5.0.
- 0.273 mL containing 3 mg lonapegsomatropin-tcgd, succinic acid (0.32 mg), trehalose dihydrate (22.7 mg) and tromethamine for pH adjustment to 5.0.
- 0.327 mL containing 3.6 mg lonapegsomatropin-tcgd, succinic acid (0.39 mg), trehalose dihydrate (27.1 mg) and tromethamine for pH adjustment to 5.0.
- 0.391 mL containing 4.3 mg lonapegsomatropin-tcgd, succinic acid (0.46 mg) and trehalose dihydrate (32.5 mg) and tromethamine for pH adjustment to 5.0.
- 0.473 mL containing 5.2 mg lonapegsomatropin-tcgd, succinic acid (0.56 mg) and trehalose dihydrate (39.3 mg) and tromethamine for pH adjustment to 5.0.
- 0.286 mL containing 6.3 mg lonapegsomatropin-tcgd, succinic acid (0.34 mg) and trehalose dihydrate (21.2 mg) and tromethamine for pH adjustment to 5.0.
- 0.345 mL containing 7.6 mg lonapegsomatropin-tcgd, succinic acid (0.41 mg) and trehalose dihydrate (25.5 mg) and tromethamine for pH adjustment to 5.0.
- 0.414 mL containing 9.1 mg lonapegsomatropin-tcgd, succinic acid (0.49 mg) and trehalose dihydrate (30.6 mg) and tromethamine for pH adjustment to 5.0.
- 0.5 mL containing 11 mg lonapegsomatropin-tcgd, succinic acid (0.59 mg) and trehalose dihydrate (37 mg) and tromethamine for pH adjustment to 5.0.
- 0.605 mL containing 13.3 mg lonapegsomatropin-tcgd, succinic acid (0.71 mg) and trehalose dihydrate (44.8 mg) and tromethamine for pH adjustment to 5.0.
CLINICAL PHARMACOLOGY
Mechanism of Action
SKYTROFA is a pegylated human growth hormone (somatropin) for once-weekly subcutaneous injection [see Clinical Pharmacology (12.3) ] .
Somatropin binds to the growth hormone (GH) receptor in the cell membrane of target cells resulting in intracellular signal transduction and a host of pharmacodynamic effects. Somatropin has direct tissue and metabolic effects, and indirect effects mediated by insulin-like growth factor-1 (IGF-1), including stimulation of chondrocyte differentiation and proliferation, stimulation of hepatic glucose output, protein synthesis and lipolysis. Somatropin stimulates skeletal growth in pediatric patients with growth hormone deficiency (GHD) as a result of effects on the growth plates (epiphyses) of long bones.
Pharmacodynamics
Somatropin released from SKYTROFA produces a linear IGF-1 dose response. In pediatric patients with GHD, with a dose change of 0.02 mg/kg/week on average resulting in a mean change in weekly average IGF-1 standard deviation score (SDS) of 0.17. In adults with GHD, a dose change of 1 mg/week results in a mean change in weekly average IGF-1 SDS of 0.7 (range, 0.3 to 1.2).
At steady state, IGF-1 levels peak approximately 2 days post-dose, with the weekly average IGF-1 occurring approximately 4.5 days post-dose. IGF-1 levels are in the normal range for GHD patients for the majority of the week, similar to daily somatropin.
Pharmacokinetics
Absorption
Following subcutaneous dose administration, SKYTROFA releases fully active somatropin via autocleavage of the TransCon linker that follows first-order kinetics.
In pediatric patients with GHD, following subcutaneous dose administration of 0.24 mg/kg/week SKYTROFA, the observed mean (CV%) steady-state peak serum concentration (C max ) of lonapegsomatropin-tcgd was 1230 (86.3) ng hGH/mL, and the median time to reach maximum concentrations (T max ) was 25 hours. For released somatropin, C max was 15.2 (83.4) ng/mL with a median T max of 12 hours. The mean (CV%) somatropin exposure over the one-week dose interval (area under the curve) was 500 (83.8) h•ng/mL. Following subcutaneous dose administration of 0.24 mg/kg/week SKYTROFA, mean C max (CV%) of the methoxypolyethylene glycol carrier was 13.1 (28.1) mcg/mL with a median T max of 36 hours.
In adult patients with GHD, following subcutaneous administration of 6.3 mg/week SKYTROFA, the estimated median steady-state C max of lonapegsomatropin-tcgd was 69.3 ng hGH/mL, and the T max was 31 hours. For released somatropin, the estimated median C max was 2.47 ng/mL with a median T max of 13 hours. The median somatropin exposure over the one-week dose interval (area under the curve) was 75.4 h•ng/mL. Following subcutaneous dose administration of 6.3 mg/week SKYTROFA, the estimated median C max of the methoxypolyethylene glycol carrier was 4.60 mcg/mL with a median T max of 34 hours.
No significant accumulation of lonapegsomatropin-tcgd and somatropin following repeat dose administration was observed.
Distribution
In pediatric patients with GHD, the mean (CV%) steady-state apparent volume of distribution of lonapegsomatropin-tcgd after subcutaneous administration of 0.24 mg/kg/week SKYTROFA was 0.13 (109) L/kg.
In adult patients with GHD, the median steady-state apparent volume of distribution of lonapegsomatropin-tcgd after subcutaneous administration of 6.3 mg/week SKYTROFA was 0.13 L/kg.
A similar distribution pattern as observed for daily somatropin is expected once somatropin is released from lonapegsomatropin-tcgd.
Elimination
Metabolism
The metabolism of somatropin involves protein catabolism in both the liver and kidneys. The methoxypolyethylene glycol carrier is cleared by the kidneys.
Excretion
In pediatric patients with GHD, the mean (CV%) lonapegsomatropin-tcgd apparent clearance at steady state was 3.2 (67) mL/h/kg following subcutaneous administration of 0.24 mg/kg/week SKYTROFA with a mean (±SD) observed half-life of 30.7 (±12.7) hours.
The apparent half-life of somatropin released from lonapegsomatropin-tcgd was approximately 25 hours.
Specific Populations
Based on a population pharmacokinetic analysis, age (3.0 to 13.7 years for pediatric patients and 23 to 75 years for adult patients), sex, race, and body weight do not have clinically meaningful effects on pharmacokinetics.
Male and Female Patients — No sex-specific pharmacokinetic studies have been performed with SKYTROFA. The available literature indicates that the pharmacokinetics of somatropin are similar in men and women.
Patients with Renal or Hepatic Impairment — No specific studies have been performed with SKYTROFA.
Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of SKYTROFA or other growth hormone products.
Anti-lonapegsomatropin-tcgd antibodies were evaluated in samples collected every 3 months in phase 3 trials in pediatric patients with GHD receiving SKYTROFA. Mean duration of exposure to SKYTROFA was 70.2 weeks. Of the 304 patients with post-baseline assessments, 19 (6.3%) showed detectable binding antibodies to lonapegsomatropin-tcgd. No apparent correlation of anti-lonapegsomatropin-tcgd antibodies to adverse events or loss of efficacy was observed. No neutralizing antibodies to SKYTROFA were detected.
Of the 247 adult patients with GHD, treated with SKYTROFA and with post-baseline assessments, antibodies against lonapegsomatropin-tcgd were detected in 14 patients (5.7%, mean duration of exposure was 47.7 weeks). There was no identified clinically significant effect of anti-lonapegsomatropin-tcgd antibodies on the safety and efficacy of SKYTROFA. No neutralizing antibodies to SKYTROFA were detected.
NONCLINICAL TOXICOLOGY
Carcinogenicity, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with lonapegsomatropin-tcgd.
Lonapegsomatropin-tcgd was not mutagenic in the Ames test, in the human chromosomal aberration assay or in the rat bone marrow micronucleus test.
In an animal fertility study, lonapegsomatropin-tcgd was administered via subcutaneous injection to male and female rats before cohabitation, through mating to implantation.
Lonapegsomatropin-tcgd did not affect fertility or early embryo-fetal development at doses up to 20-fold the clinical pediatric dose of 0.24 mg/kg/week and approximately 46-fold the maximum clinical therapeutic dose for adult GHD of 6.3 mg hGH/week.
CLINICAL STUDIES
Treatment-Naïve Pediatric Patients With Growth Hormone Deficiency (NCT02781727)
A multi-center randomized, open-label, active-controlled, parallel-group phase 3 study was conducted in 161 treatment-naïve, prepubertal pediatric subjects with growth hormone deficiency (GHD); 105 subjects received once-weekly SKYTROFA, and 56 received daily somatropin. The dose in both arms was 0.24 mg/kg/week. The primary efficacy endpoint was annualized height velocity at Week 52.
The subjects ranged in age from 3.2 to 13.1 years with a mean of 8.5 years. One hundred thirty-two (82%) subjects were male and 29 (18%) were female. One subject was Asian, three were Black or African American, 152 were Caucasian, and five were categorized as "other." The subjects had a mean baseline height SDS (standard deviation score) of -2.9.
Treatment with once-weekly SKYTROFA for 52 weeks resulted in an annualized height velocity of 11.2 cm/year. Subjects treated with daily somatropin achieved an annualized height velocity of 10.3 cm/year after 52 weeks of treatment. Refer to Table 5.
| Once-Weekly SKYTROFA (N = 105) | Daily Somatropin (N = 56) | Estimate of Treatment Difference (95% CI) (SKYTROFA minus Daily Somatropin) | |
|---|---|---|---|
| Annualized Height Velocity (cm/year) The estimates of least squares (LS) means and 95% confidence interval (CI) are from an ANCOVA model that included baseline age, peak GH levels (log transformed) at stimulation test, baseline height SDS – average SDS of parental height as covariates, and treatment and sex as factors. Missing data were imputed with multiple imputation method. | 11.2 | 10.3 | 0.9 (0.2-1.5) |
Height SDS (change from baseline) was 1.1 in the SKYTROFA arm and 0.96 in the daily somatropin arm at Week 52. Refer to Table 6.
| Once-Weekly SKYTROFA (N = 105) | Daily Somatropin (N = 56) | |
|---|---|---|
| Abbreviations: SDS, standard deviation score. | ||
| Height SDS, baseline | -2.9 | -3.0 |
| Height SDS, change from baseline Height SDS, change from baseline: The estimates of LS means are from an ANCOVA model that included baseline age, peak GH levels (log transformed) at stimulation test and baseline height SDS as covariates, and treatment and sex as factors. | 1.1 | 0.96 |
Adults With Growth Hormone Deficiency (NCT05171855)
A multi-center, randomized, parallel-group, placebo-controlled (double-blind), phase 3 study was conducted in 259 adults with GHD. Eighty-nine subjects received once-weekly SKYTROFA, 84 subjects received once-weekly placebo, and 86 subjects received open-label daily somatropin.
The mean (range) age at enrollment was 43 (23 to 81) years old, with 119 (46%) females (55 on oral estrogen) and 140 (54%) males, and the (SD) baseline body mass index of 28 (6.3) kg/m 2 . One subject was American Indian or Alaska Native, 1 was Black or African American, 28 were Asian, and 218 were Caucasian.
The primary efficacy endpoint, change in trunk percent (%) fat was measured by dual X-ray absorptiometry from baseline to Week 38 in the SKYTROFA group, compared to the placebo group (see Table 7 ).
| Change from Baseline at Week 38 | Once-Weekly SKYTROFA (N = 89) | Once-Weekly Placebo (N = 84) | LS Mean Difference [95% CI] | P-value |
|---|---|---|---|---|
| Abbreviations: LS, least squares; CI, confidence interval; kg, kilogram. | ||||
| Trunk percent fat (%) The estimates of least squares (LS) means and 95% confidence interval (CI) are from an ANCOVA model that included treatment arm, region (North America, Europe, Asia-Pacific), baseline age group, gender, concomitant oral estrogen at screening (yes vs. no), and adult GHD onset (adult vs. childhood) and corresponding baseline variable as a covariate. Missing data were imputed with multiple imputation method. | -1.7 | 0.4 | -2.0 [-2.9, -1.1] | < 0.0001 |
Patients treated with daily somatropin achieved a change in trunk percent fat of -3.1% after 38 weeks. No formal statistical comparison between SKYTROFA and daily somatropin was conducted.
Change in total body lean mass and trunk fat mass from baseline after 38 weeks of treatment were secondary efficacy endpoints.
At 38 weeks, the change from baseline in total body lean mass was +1.6 kg for lonapegsomatropin and -0.1 kg for placebo (LS mean difference of 1.7 kg with 95% CI of 1.0 to 2.5, p-value < 0.0001).
At 38 weeks, the change from baseline in trunk fat mass was -0.5 kg for lonapegsomatropin and +0.2 kg for placebo (LS mean difference of -0.7 kg with 95% CI of -1.2 to -0.2, p-value = 0.005).
After 38 weeks, SKYTROFA treatment in adults with GHD resulted in normalization of IGF-1 SDS to 1.4 compared to -2.6 in placebo-treated patients. See Table 8 .
| Time Point | Once-Weekly SKYTROFA (N = 89) | Once-Weekly Placebo (N = 84) |
|---|---|---|
| Abbreviations: SD, standard deviation; SDS, standard deviation score. | ||
| Baseline IGF-1 SDS, mean (SD) | -2.6 (1.0) | -2.7 (1.2) |
| Week 38 IGF-1 SDS, mean (SD) Sampling time corresponded to weekly average IGF-1 SDS for SKYTROFA. | 1.4 (1.9) | -2.6 (1.3) |
The mean (SD) IGF-1 SDS level in daily somatropin treated patients was -2.82 (1) at baseline and 0.49 (1.98) at 38 weeks. No formal statistical comparison between SKYTROFA and daily somatropin was conducted.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
SKYTROFA (lonapegsomatropin-tcgd) for injection is a sterile, preservative-free, white to off-white lyophilized powder available in a single-dose, dual-chamber, prefilled cartridge containing lonapegsomatropin-tcgd in one chamber and the diluent, Water for Injection, in the second chamber. The dual-chamber glass cartridge is available in 14 strengths (in somatropin equivalents) as described in Table 9.
| SKYTROFA | NDC |
|---|---|
| 0.7 mg | 73362-012-01 |
| 1.4 mg | 73362-013-01 |
| 1.8 mg | 73362-014-01 |
| 2.1 mg | 73362-015-01 |
| 2.5 mg | 73362-016-01 |
| 3 mg | 73362-003-01 |
| 3.6 mg | 73362-004-01 |
| 4.3 mg | 73362-005-01 |
| 5.2 mg | 73362-006-01 |
| 6.3 mg | 73362-007-01 |
| 7.6 mg | 73362-008-01 |
| 9.1 mg | 73362-009-01 |
| 11 mg | 73362-010-01 |
| 13.3 mg | 73362-011-01 |
Each carton contains 4 single-dose prefilled cartridges and 6 sterile, single-use, disposable 0.25 mm × 4 mm (31-gauge × 5/32 inch) needles. The cartridges are for use only with the SKYTROFA Auto-Injector, packaged in a separate carton. The SKYTROFA Auto-Injector is not supplied with SKYTROFA cartridges but is available for patients with a prescription for SKYTROFA through the Ascendis Pharma Customer Support by calling the toll-free number at 1-844-442-7236 (1-844-44ASCENDIS).
Storage and Handling
- For patients : Refrigerate SKYTROFA cartridges at 36°F to 46°F (2°C to 8°C) in the outer carton to protect from light until the expiration date. Do not freeze. Alternatively, SKYTROFA outer carton containing blistered cartridges may be stored at room temperature [up to 86°F (30°C)] for up to 6 months and can be returned to refrigeration within the 6 months. Write the date first removed from the refrigerator in the space provided on the outer carton. Do not use SKYTROFA beyond the expiration date or 6 months after the date it was first removed from refrigeration (whichever is earlier).
- For pharmacy long-term storage : Store SKYTROFA cartridges refrigerated at 36°F to 46°F (2°C to 8°C) in the outer carton to protect from light until the expiration date. Do not freeze.
| Keep Track of Your Injections | This is your Instructions for Use | Instructions for Use |
 |  |  |
- Important Information
- Before You Begin
- Setting Up
Step-by-Step Guide
- Troubleshooting
- Cleaning and Maintenance
- Charging and Charging Cable
- Storing
- Product Safety
- Expiration
- EMC Compliance Levels
- Technical Specification
- Symbols
- Warranty and Disclaimer
- Parts Overview
Introduction
Intended Purpose and Device Description:
The SKYTROFA Auto-Injector is intended to automate the mixing (reconstitution) and injection under the skin (subcutaneous use) of lonapegsomatropin in patients prescribed one-time weekly growth hormone therapy.
Injections are given under the skin (subcutaneously) one-time weekly in the abdomen, thighs, or buttocks.
The intended users are patients prescribed with SKYTROFA, caregivers, and healthcare providers.
Always use SKYTROFA exactly as your healthcare provider has told you. If you are unsure about your medicine, contact your healthcare provider.
Consult your healthcare provider to be advised to perform self-administration or administration by your caregiver.
The auto-injector is an electromechanical, reusable, single-patient use, and single-dose delivery device designed for personal home use. This means that a patient or caregiver can give injections at home without a healthcare provider present.
A healthcare provider can use the auto-injector in a non-emergency clinical setting to inject a patient or demonstrate device use.
Contraindications and Side Effects
For more information on SKYTROFA contraindications, side effects, disease, self-care, summary of safety, and clinical performance, please refer to the information enclosed with your medicine or contact your healthcare provider.
If you get any side effects, contact your healthcare provider.
Step-by-step Instructions for the SKYTROFA Auto-Injector
Read and follow this Instructions for Use that comes with your auto-injector before you start using it. This information does not replace talking to your healthcare provider about your medical condition or your treatment.
The back cover of this Instructions for Use folds out for reference while you read the rest of the instructions.
If you have any questions about the auto-injector, the medicine, or these instructions, please contact your healthcare provider or Ascendis Pharma Customer Support. For contact information 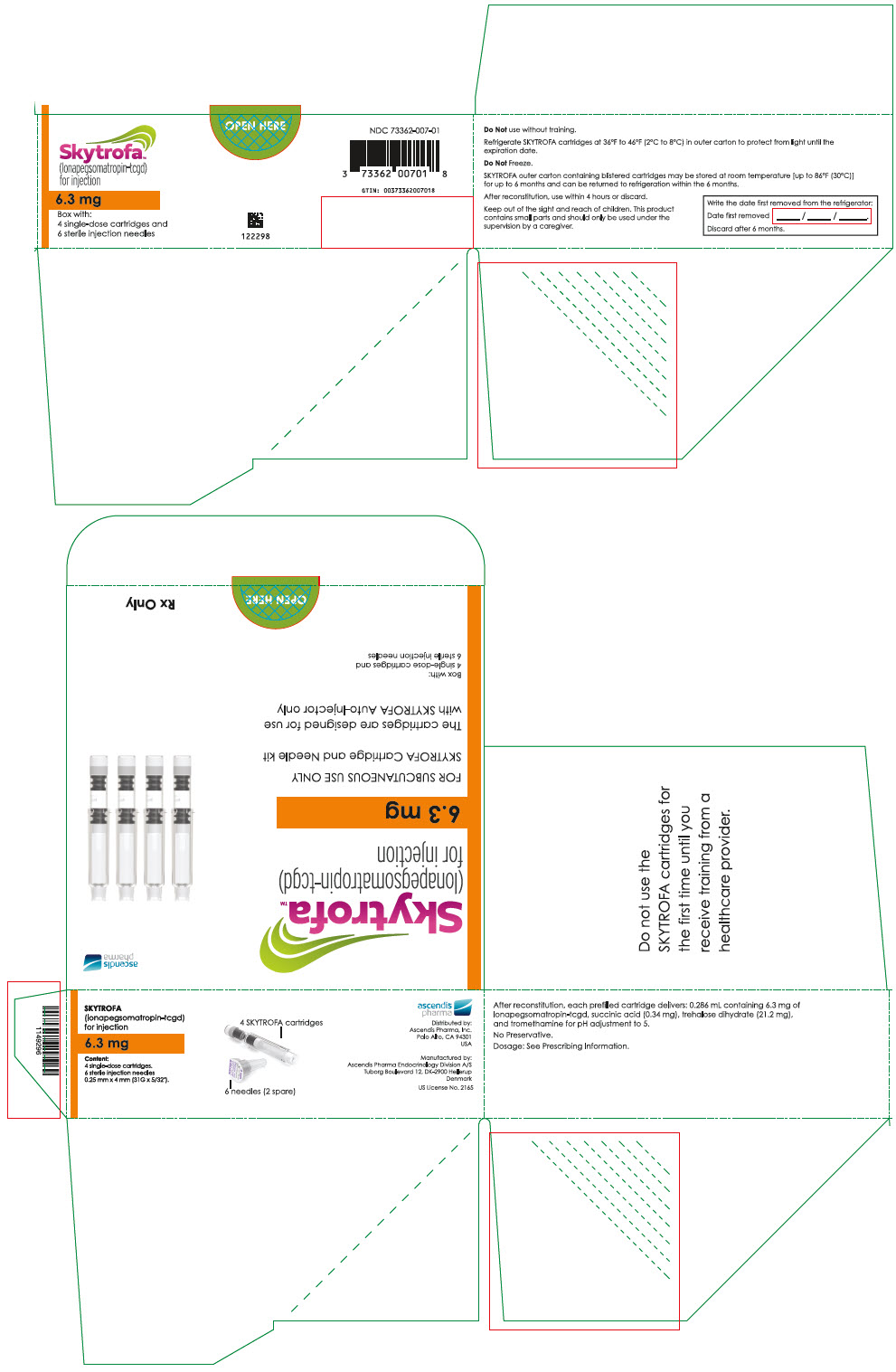 back cover.
back cover.
Getting Started
| Page | |
|---|---|
| Important Information | 6 |
| Before You Begin | 9 |
| Setting Up | 12 |
Important Information 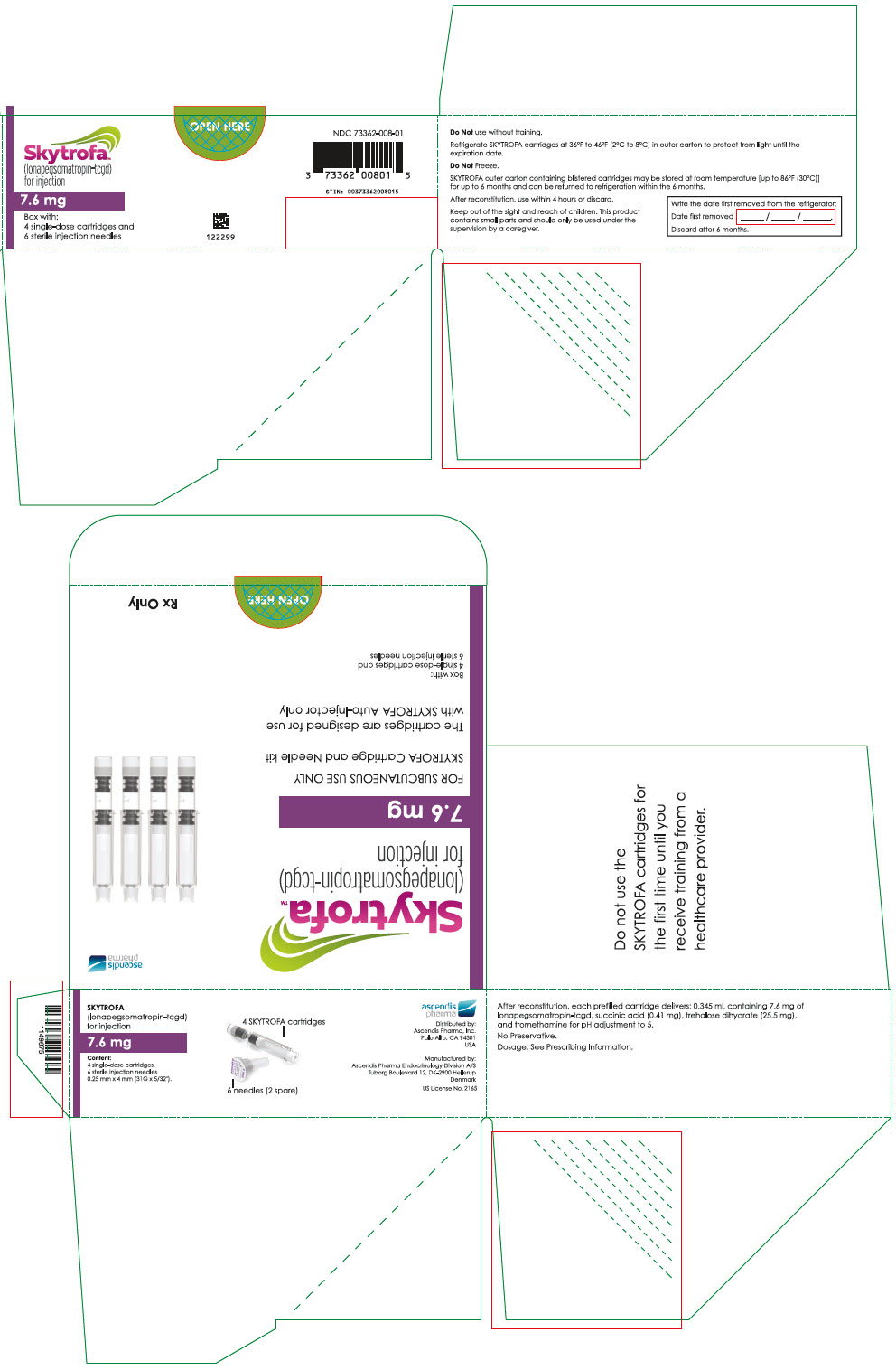
Important information about your SKYTROFA Auto-Injector:
Do not use the SKYTROFA Auto-Injector for the first time until you receive training from a healthcare provider. Follow this Instructions for Use for the auto-injector to avoid getting an injury, infection, or incorrect dose.
The auto-injector can only be used with SKYTROFA Cartridges and needles that are prescribed by your healthcare provider. Cartridges and needles come together in the same packaging . Follow the instructions that come with SKYTROFA Cartridges. If refrigerated, take the cartridge out of the refrigerator and leave at room temperature for 15 minutes before use.
Do not use your auto-injector with other medicines or needles than provided in the SKYTROFA Cartridge pack.
 | Your weekly dose may require that you use 2 cartridges. |
 | When this warning symbol is seen, safety instructions must be followed. Otherwise, serious safety hazardous situations and injuries may occur. |
 | Do not share your auto-injector with other people, even if the needle has been changed. You may give other people a serious infection or get a serious infection from them. |
 | Keep out of the reach of children. This product contains small parts that may present a choking hazard to small children. The cable can present a strangulation hazard. |
 | Do not use any charger or charging cables other than those included with your auto-injector. Read more on page 65. |
 | Do not use or place the auto-injector closer than 12 inches (30 cm) to microwave ovens or electronic equipment with antennas such as Wi-Fi transceivers. The auto-injector might not work the right way. |
 | When this precaution symbol is seen, safety instructions must be followed. Otherwise, safety hazardous situations and injuries and potential damage to the auto-injector or other property may occur. |
 | Do not reuse needles. |
 | Do not use a cartridge if it has been dropped or you think it may be damaged. |
 | Do not point the auto-injector at yourself or other people, except when you are ready to inject, to avoid needle stick injury. |
 | If your auto-injector turns off automatically, press and release the green button to turn it on again and read page 60 to proceed. |
 | If the auto-injector packaging has been damaged or unintentionally opened before first using it, please call Ascendis Pharma Customer Support. For contact information |
 back cover.
back cover. Before You Begin
The SKYTROFA Auto-InjectorYour healthcare provider will inform you that the SKYTROFA Auto-Injector, which comes in a separate package, is required for injecting the prescribed the SKYTROFA medicine.
The SKYTROFA Auto-Injector is exclusively for use with SKYTROFA Cartridge (medicine).
The SKYTROFA Auto-Injector automates part of the procedure for injecting SKYTROFA medicine.
The SKYTROFA Cartridge
The medicine SKYTROFA comes in a one-time use cartridge "SKYTROFA Cartridge". The cartridge has 2 chambers, 1 filled with powder and 1 filled with water. The auto-injector automatically mixes the powder and the water during mixing steps, making it ready for injection. Each SKYTROFA Cartridge pack contains 4 cartridges packed in individual blisters.
The Needle
The single-use needle comes with SKYTROFA Cartridges and is used for screwing on the cartridge to inject the medicine. Each SKYTROFA Cartridge pack contains 6 needles including 2 spare needles.
Contact your healthcare provider if you need more SKYTROFA consumables.
 Figure A
Figure A
Product Overview
 Figure B
Figure B
Setting Up
- 1.000000000000000e+00 Remove the SKYTROFA Auto-Injector from the package.
- Use the auto-injector at room temperature between 59°F to 86°F (15°C to 30°C) .
- 2.000000000000000e+00 Connect the USB (large end of the cable) to the charger (only use the provided charger). Plug the charger into a power outlet. Connect the Micro-USB (small end of the cable) to the back of the auto-injector. For protection, charge the auto-injector with the protective cover on (see Figure C ).
For more information on charging
 page 65.
page 65.  Figure C
Figure C
- 3.000000000000000e+00 Fully charge the auto-injector before using it for the first time . This will take 2 hours and 30 minutes. When the battery icon located at the base of the auto-injector (see Figure D ) is flashing orange or green, it is charging. When it shows constant green, the auto-injector is fully charged and is ready to use after the charging cable is unplugged.
- When fully charged, the battery should last for at least 3 injections.
- When traveling, bring regional adaptors for the auto-injector charger to connect to the local power outlet.
The auto-injector cannot be used when connected to the charger.
 Figure D
Figure D
- 4.000000000000000e+00 Find a quiet place where you can perform your injection.
- 5.000000000000000e+00 Gather your supplies and place them on a flat, hard, clean surface. Supplies needed for an injection: From SKYTROFA Auto-Injector packaging (see Figure B ):
- 1 SKYTROFA Auto-Injector
- 1 SKYTROFA Cartridge
- 1 Omnican fine 0.25 mm × 4 mm (31G × 5/32") B. Braun needle
- 1 Alcohol Wipe
- 1 Sharps Container
 If your weekly dose requires 2 cartridges, you will need the following additional supplies:
If your weekly dose requires 2 cartridges, you will need the following additional supplies:
- 1 SKYTROFA Cartridge
- 1 Omnican fine 0.25 mm × 4 mm (31G × 5/32") B. Braun needle
- 1 Alcohol Wipe
Supplies Needed for Each Injection
 Figure E
Figure E
|
Your healthcare provider may prescribe a dose that requires use of the medicine in 2 cartridges. If you have been prescribed a dose that requires 2 cartridges:
|
| Figure F |

 Does Your Weekly Dose Require 2 Cartridges?
Does Your Weekly Dose Require 2 Cartridges? How to Use
| Step-by-Step Guide | Page |
|---|---|
| Prepare | 20 |
| Mix | 30 |
| Inject | 38 |
| After Injection | 44 |
 | |
Prepare
1 Check and assemble cartridge and needle
 Figure G
Figure G
| 1.1 | Check the expiration date and cartridge dose on the cartridge packaging (see Figure G ). |
 | Do not use if the dose is not as prescribed. |
| Remove the SKYTROFA Cartridge from the packaging according to the instructions on its lid. | |
| If refrigerated, take the cartridge out of the refrigerator and leave at room temperature for 15 minutes before use. | |
Do not use if the expiration date has passed on the cartridge.
| |
| 1.2 | Check the expiration date on the needle. Peel off the paper from the needle by hand (see Figure H ). |
| |
| |
| |
| 1.3 | Screw the needle straight on the cartridge by turning clockwise until there is a tight fit (see Figure I and J ). |
 | Do not remove the plastic needle cover. You will need it to insert the cartridge into the SKYTROFA Auto-Injector. Check the needle is screwed tight on the cartridge. |
 | Do not use the needle if it has touched anything other than the injection site. |
| |
| Check the needle is screwed tight on the cartridge (see Figure J ). | |
| |
| 2 Turn on the auto-injector | |
| |
| 2.1 | Disconnect the auto-injector from the charger when charged.
|
| 2.2 | Find a quiet place where you can give your injection. Use the auto-injector at room temperature between 59°F to 86°F (15°C to 30°C). |
| 2.3 | Remove the protective cover. Place the auto-injector upright on a flat surface. |
| 2.4 | Press and release the green button to turn on the auto-injector (see Figure K ).
|
| 2.5 | Check the battery icon on the base of the auto-injector to see if it is charged. The battery icon |
 | Constant Green Battery is fully charged. The auto-injector is ready to use. |
 | Flashing Green At least 1 injection remaining, but charging is recommended after use. |
 | Flashing Orange Battery needs charging. |
If no icons light up | |
If you see flashing icons (other than the battery) | |
 back cover.
back cover.  Be careful with the exposed needle to avoid needle stick injury and infection.
Be careful with the exposed needle to avoid needle stick injury and infection.  Do not use the needle if:
Do not use the needle if:  Do not use a bent needle.
Do not use a bent needle.  Figure H
Figure H  Figure I
Figure I  Figure J
Figure J  Figure K
Figure K  and the 3 icons above the green button will light up. Then all of the icons will turn off, except the battery icon.
and the 3 icons above the green button will light up. Then all of the icons will turn off, except the battery icon.  is green when the auto-injector is ready to use:
is green when the auto-injector is ready to use:  page 56.
page 56.  pages 58–59.
pages 58–59. | 3 Insert cartridge with attached needle | |
| |
| 3.1 | Insert the cartridge. |
 | Insert the cartridge into the flashing green top by pushing straight down with the needle cover still on (see Figure L ). |
| 3.2 | Click the cartridge into place. |
 | Make sure the cartridge is pushed all the way down until you hear a click (see Figure M ).
|
 | Do not insert the cartridge without attaching the needle fully. If you did not attach the needle, push the cartridge to release it and continue with Step 1.2 (page 21). |
 | Do not attempt to insert the charging cable in the auto-injector if a cartridge has been inserted. |
| 3.3 | After the click, remove your finger from the cartridge (see Figure N ).
|
 | If your auto-injector turns off automatically, press and release the green button to turn it on again, then read Situation A |
 | If you see a flashing orange mixing icon |
| |
Do not use the auto-injector if you cannot insert the cartridge. Please call Ascendis Pharma Customer Support. For contact information | |

 will light up, and the battery icon
will light up, and the battery icon  will switch off.
will switch off.  page 61.
page 61.  page 58. If you cannot insert the cartridge, check if an orange plug is still attached to the cartridge. If an orange plug is still attached to the cartridge, remove it by pulling it straight off (see
page 58. If you cannot insert the cartridge, check if an orange plug is still attached to the cartridge. If an orange plug is still attached to the cartridge, remove it by pulling it straight off (see  Figure O
Figure O  back cover.
back cover. Mix
| 4 Wait while mixing | |
| |
| 4.1 | Wait 4 to 8 minutes for the auto-injector to mix your medicine. The progress bar will gradually light up, and you will hear steady ticking during mixing (see Figure P ).
|
 | If you see a slowly flashing green mixing icon and the progress bar is frozen |
 | If you see a flashing orange mixing icon |
| 4.2 | The auto-injector has finished the automatic part of the mixing when you hear 2 loud beeps and the entire progress bar flashes. |
| 4.3 | Continue with Step 5 (page 32) immediately after automatic mixing is completed. If you wait for more than 2 hours before completing the steps for mixing the medicine by hand (Step 5), the auto-injector will automatically cancel the procedure. If this happens, the cartridge will be released and cannot be used. To remove the cartridge, see Step 10 (page 44). If you still need to inject after the auto-injector has canceled the procedure, go back to Step 1 and use a new cartridge (page 20). |
 | If your auto-injector turns off automatically, press and release the green button to turn it on again, then read Situation B |
 Figure P
Figure P  page 54.
page 54.  page 58.
page 58.  page 61.
page 61. | 5 Turn the auto-injector up and down | |
| Turn the auto-injector up and down, listening for a tick sound each up and down cycle to make sure the turns are correct. |  |
| Turn up and down correctly 5 to 10 times until you hear 2 loud beeps and the whole progress bar, except the top element, lights up. | |
| Do not press the green button. | |
| Figure Q | |
| 5.1 | Turn the auto-injector up and down to mix the medicine by hand. You will hear a 'tick' sound each time you turn the auto-injector up and down correctly. To mix the medicine correctly:
|
 | If your auto-injector turns off automatically, press and release the green button to turn it on again. Read Situation B or C to find the corresponding lit-up icons |
 | If you see a flashing orange mixing icon |
| 5.2 | Continue with Step 6 (page 36) immediately after you finish mixing by hand. If you wait more than 2 hours before performing Step 6 to Step 9 (pages 36–43), the auto-injector will automatically cancel the procedure. If this happens, the cartridge will be released and cannot be used. To remove the cartridge, see Step 10 (page 44).
|
 page 61 and 62.
page 61 and 62.  page 58.
page 58. | 6 Finish mixing | |
| |
| 6.1 | Keep the auto-injector upright for automatic air removal (see Figure R ). Wait until you hear 2 loud beeps and the entire progress bar lights up. |
 | If you see a slowly flashing green mixing icon and the progress bar is frozen |
 | If you see a flashing orange mixing icon |
| 6.2 | Pull off the needle cover (see Figure S ). The green eye icon |
 | Keep the needle cover for later use . It is needed to safely remove the cartridge after injection. |
 | If your auto-injector turns off automatically, press and release the green button to turn it on again. Read Situation D or E to find the corresponding lit-up icons |
| Inject | |
| 7 Check mixed medicine | |
| |
| 7.1 | Check the mixed medicine in the inspection window on the side of the auto-injector (see Figure T ). The medicine should look colorless and clear. It may contain air bubbles. This is normal. |
 | Be careful with the exposed needle to avoid needle stick injury and infection. Do not inject the medicine if there are visible particles (medicine is not dissolved) or the mixed medicine is discolored. If you see visible particles or the medicine is discolored:
|
 | If you see a flashing orange mixing icon |
| 8 Prepare for injection | |
| |
| 8.1 | Choose an injection site. |
 | There are only 3 areas of your body you can inject into (see Figure U ):
|
| 8.2 | Make sure your hands are clean using soap and water or hand sanitizer (see Figure V ). |
| |
| 8.3 | Clean the injection site with an alcohol wipe (see Figure W ). |
 | Do not inject in an injection site that has not been cleaned. |
 | Do not touch the cleaned area before injecting. |
 | Do not fan or blow on the cleaned area. |
 | Inject directly into skin. Do not inject through clothes. |
| |
| 9 Inject medicine | |
| |
| 9.1 | Press and hold the green top against the skin of the injection site to inject (see Figure X ). Hold for 10 to 20 seconds until you hear 2 loud beeps and the green top flashes 2 times. The green check mark icon
|
 | Do not remove the auto-injector from the injection site until the injection is finished to ensure you get your full cartridge dose. The injection is finished when you hear 2 loud beeps and the green top flashes 2 times. The green check mark |
| 9.2 | Remove the auto-injector from the skin after the injection is finished (see Figure Y ).
|
 | If you see a slowly flashing green check mark icon and the progress bar is frozen |
 | If you see a flashing orange check mark icon |
| After Injection | |
| 10 Remove cartridge | |
| |
 | Be careful when handling needles to reduce the risk of needlestick injury and infection. |
| 10.1 | Press down the needle cover into the flashing green top until you hear a click to remove the cartridge (see Figure Z ).
|
| 10.2 | Remove the used cartridge by pulling straight up (see Figure AA ). |
 | Do not remove the cartridge without using the needle cover.
|
 | Do not use the auto-injector if you cannot remove the cartridge as instructed. Please call Ascendis Pharma Customer Support. For contact information |
| 11 Check cartridge and throw away | |
| |
| 11.1 | Check that the cartridge is empty of medicine (see Figure AB ). |
 | Do not use the auto-injector if there is medicine left in the cartridge after injection. Please call Ascendis Pharma Customer Support. For contact information |
| 11.2 | Put your used cartridge and needle in an FDA-cleared sharps disposal container right away after use (see Figure AB ). |
 | Do not throw away (dispose of) opened or used needles and cartridges in your household trash. Do not recycle your used sharps disposal container. If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
|
 | Healthcare providers, relatives, and other caregivers should follow this Instructions for Use for removal and throwing away (disposal) of needles to prevent needlestick injury and infection. |
 | Does Your Weekly Dose Require 2 Cartridges? Then take the second injection by repeating Step 1 to Step 11 (pages 20–47) with a new cartridge and needle before continuing to Step 12. |
| 12 Store the auto-injector | |
| |
| 12.1 | Make sure that the auto-injector is clean. If it is dirty or if medicine has been spilled onto it, clean with a damp cloth and wait until dry . Do not place the auto-injector under water. For more information on cleaning |
| 12.2 | Put the protective cover on the auto-injector by sliding it straight down (see Figure AC ). |
| 12.3 | Charge the auto-injector if the battery icon has been flashing before or after the injection. For more information about how to charge the auto-injector |
| 12.4 | It is recommended to store the auto-injector at room temperature between 59°F to 86°F (15°C to 30°C), in between uses. Store with the protective cover on until the next injection. For more information about how to store the auto-injector |
| 12.5 | Write down the date of every weekly dose taken under Keep Track of Your Injections
|

 page 54.
page 54.  page 58.
page 58.  will light up. Removing the needle cover will allow you to check the mixed medicine in the inspection window (Step 7 on page 38). Do not twist the needle cover off. If you have trouble removing the needle cover, gently pull up the green top.
will light up. Removing the needle cover will allow you to check the mixed medicine in the inspection window (Step 7 on page 38). Do not twist the needle cover off. If you have trouble removing the needle cover, gently pull up the green top.  page 62 and 63.
page 62 and 63.  Figure T
Figure T  page 58.
page 58.  Figure U
Figure U  Figure V
Figure V  Figure W
Figure W 
 will light up.
will light up.  will light up.
will light up.  page 55.
page 55.  page 58.
page 58. 
 will display the battery level. The auto-injector turns off automatically.
will display the battery level. The auto-injector turns off automatically.  back cover.
back cover.  Figure AB
Figure AB  back cover.
back cover. 
 page 64.
page 64.  page 65.
page 65.  page 66.
page 66.  back cover fold out.
back cover fold out.  page 72.
page 72. Troubleshooting and Care
| Page | |
|---|---|
| Troubleshooting | 52 |
| Cleaning and Maintenance | 64 |
| Charging and Charging Cable | 65 |
| Storing | 66 |
Troubleshooting
- What do you see?

| Orange flashing icons If you see any of these |
 | Orange battery icon If you see this |
If the auto-injector turns off automatically, press and release the green button: If the auto-injector is not responding with no icons lighting up |



 pages 58–59.
pages 58–59.  page 57.
page 57.  page 56. If any progress bar elements or icons light up
page 56. If any progress bar elements or icons light up  page 61–63.
page 61–63.
| You see slowly flashing green mixing icon and the progress bar is frozen. |
 | You hear a repeating notification sound. |
| Mixing paused (Step 4 on page 30 or Step 6 on page 36). If the SKYTROFA Auto-Injector is not in an upright position, mixing or air removal will pause (see Figure AD ). | |
| Do this: Place the auto-injector in an upright position and the mixing or air removal will continue. | |
| |
| You see a slowly flashing green check mark icon and the progress bar is frozen. |
 | You hear a repeating notification sound. |
| Injection paused (Step 9 on page 42). If the green top is removed from the skin before completing the injection, the auto-injector will pause the injection (see Figure AE ). | |
| Do this: Press the green top against skin and the injection will start again. If the injection is not completed within 15 minutes, the auto-injector automatically cancels the injection. | |
| |
 | You see a flashing green battery icon . |
| Charging recommended. There is enough battery for at least 1 injection. | |
| Do this: Charge the auto-injector after the next injection. | |
 | You see a constant green battery icon . |
| Battery is fully charged. The auto-injector is ready to use (the battery icon turns off while the auto-injector is in use). | |
| Do this: If you are charging the auto-injector, disconnect it from the charger. | |
| You see no icons lighting up at all. | |
| Either the auto-injector has turned off (the auto-injector will turn off if it is not active for 6 minutes without a cartridge loaded or for 3 minutes with a cartridge loaded) or the battery needs charging. | |
Do this: Press the green button to turn it on again.
| |
 | You see a flashing orange battery icon . You hear a notification sound. |
| The battery needs charging. | |
| Do this: Charge the auto-injector. | |
| Insert the charging cable into the auto-injector at rear lower side and connect to a power outlet (see Figure AF ). When the auto-injector has charged for 15 minutes and the battery icon flashes green, the auto-injector is ready to use for at least 1 injection after the charging cable is unplugged. When the battery icon shows constant green, the auto-injector is fully charged and lasts for at least 3 injections. | |
| |
| You see a flashing orange mixing icon . |
 | You hear an error sound. |
| Either you have inserted a used cartridge, or you have canceled the mixing, or the auto-injector has canceled the injection, or the cartridge is damaged. The auto-injector cancels the injection 2 hours after automatic mixing (Step 4, page 30) or 2 hours after manual mixing (Step 5, page 32) is completed. | |
Do this: Remove the cartridge and wait for the device to turn off. Start again with a new, unused cartridge and make sure to screw on the needle straight and tightly. Read more | |
| You see a flashing orange check mark icon . |
 | You hear an error sound. |
| Either an injection error has happened (the needle is not screwed on tightly, or it is bent or blocked), or you have canceled the injection, or the auto-injector has canceled an injection that was not completed within 15 minutes. | |
Do this: Remove the cartridge and wait for the auto-injector to turn off. Start again with a new, unused cartridge and make sure to screw on the needle straight and tightly. Read more | |


 Figure AD
Figure AD 

 Figure AE
Figure AE  page 57.
page 57.  page 60.
page 60.  Figure AF
Figure AF 

 page 20.
page 20. 

 page 20.
page 20.
| You see 3 flashing icons alternating between green and orange . |
| |
| You hear a repeating error sound. |






| The auto-injector is near its expiration. The first time you see this, either there are 5 injections left or the auto-injector expires in 1 month. | |
Do this: Press and release the green button to proceed. The green top will start flashing. The auto-injector can be used as usual for the remaining injections. When there are no more injections left or the expiration date has passed, the auto-injector cannot be used. Please contact a healthcare provider or Ascendis Pharma Customer Support to receive a new auto-injector. For Ascendis Pharma contact information | |
  | You see 3 flashing orange icons . |
 | You hear an error sound. |
| Either the auto-injector has expired (the maximum number of injections has been reached or the lifetime of 5 years has passed) or a critical error has occurred. | |
Do this: Try to turn the auto-injector on (press and release the green button). If you still see 3 flashing orange icons, the auto-injector needs to be replaced. For disposal information | |
 | If you see no icons lighting up at all. Either the auto-injector needs charging or it has timed out and turned off automatically. |
 | Do this: Press and release the green button to turn the auto-injector on again. |
 |
|
Do this: Read more | |
 |
|
| Do this: Read the next pages and find the way your device lights up and blinks. | |
 | Situation A You see a flashing green mixing icon and a progress bar with the bottom first element lit up. You hear a repeating notification sound. The auto-injector has not completed the automatic medicine mixing. Do this : Keep the auto-injector standing upright on a flat surface. Go to |
 | Situation B You see a constant green mixing icon and a flashing progress bar . The auto-injector has not completed the manual mixing of the medicine. Do this: Continue to turn the auto-injector up and down to mix the medicine by hand. Go to |
 | Situation C You see a flashing green mixing icon and a constant progress bar lit up. You hear a repeating notification sound. The auto-injector has not automatically removed the air. Do this: Keep the auto-injector standing upright on a flat surface. Go to |
 | Situation D You see a constant green mixing icon and a constant progress bar lit up. You have not pulled off the needle cover within 3 minutes of completing automatic air removal. Do this: Continue to pull off the needle cover. Go to |
 | Situation E You see a constant green eye icon. You have not started to inject within 3 minutes of pulling off the needle cover. Do this: Continue to prepare and do the injection. Go to |
| Cleaning and Maintenance | |
| Cleaning the SKYTROFA Auto-Injector If the auto-injector is dirty or if medicine has been spilled onto it, clean with a damp cloth . Before cleaning, the cartridge must be removed. The green top needs to be cleaned on the outside and on the reachable part of the inside. If medicine is spilled inside the auto-injector, turn it upside down to let the medicine run out. | |
 | Do not clean the auto-injector while charging. Keep the auto-injector dry. Do not place the auto-injector in liquid. Do not sterilize the auto-injector. |
| Maintenance of the SKYTROFA Auto-Injector | |
 | Do not open, try to repair, or change the auto-injector. The battery is not replaceable. Please call Ascendis Pharma Customer Support. For contact information |
| Charging and Charging Cable | |
| Charging the SKYTROFA Auto-Injector | |
Connect the USB (large end of the cable) to the charger. Plug the charger into a power outlet. Connect the Micro-USB (small end of the cable) to the rear of the auto-injector. For protection, charge the auto-injector with the protective cover on. Read more | |
 | Do not use any charger or charging cables other than those included with your auto-injector. Other chargers or cables may create problems and may interfere with device operation. |
 | Do not clean the auto-injector while charging. Keep the auto-injector dry. |
 | Do not insert a cartridge when the auto-injector is connected to the charger. |
 | Do not attempt to insert the charging cable in the auto-injector if a cartridge has been inserted. |
| Storing | |
| Storing the SKYTROFA Auto-Injector | |
 | Keep out of the reach of children. This product contains small parts that may present a choking hazard to small children. The cable can present a strangulation hazard. |
 | Do not store the auto-injector closer than 12 inches (30 cm) to microwave ovens or electronic equipment with antennas such as Wi-Fi transceivers. The auto-injector may not work the right way. Remove the cartridge from the auto-injector before storing. Store with the protective cover on. It is recommended to store the auto-injector at room temperature between 59°F to 86°F (15°C to 30°C), in between uses. If the auto-injector is stored at higher or lower temperatures, you should keep it at room temperature for 30 minutes before use with a cartridge. The auto-injector can be stored between 14°F to 104°F (-10°C to 40°C) if not in use. Keep the protective cover on when traveling with the auto-injector. Keep the auto-injector away from dirt, dust, and humid or wet places. Keep the auto-injector, the charger, and the charging cable away from pets and pests. Avoid refrigerating or freezing the auto-injector in between uses. |
 | Avoid exposing the auto-injector to direct sunlight and extreme temperatures. For technical details of the conditions under which the auto-injector can be used, stored, and transported |
 back cover.
back cover.  page 73. Please contact a healthcare provider or Ascendis Pharma Customer Support to receive a new auto-injector. For Ascendis Pharma contact information
page 73. Please contact a healthcare provider or Ascendis Pharma Customer Support to receive a new auto-injector. For Ascendis Pharma contact information  back cover.
back cover.  page 57.
page 57.  Step 4.1 on page 30.
Step 4.1 on page 30.  Step 5.1 on page 32.
Step 5.1 on page 32.  Step 6.1 on page 36.
Step 6.1 on page 36.  Step 6.2 on page 37.
Step 6.2 on page 37.  Step 7 to Step 9, pages 38–43.
Step 7 to Step 9, pages 38–43.  back cover.
back cover.  pages 12–13. Only charge the auto-injector when the battery icon is flashing green or orange. Read more
pages 12–13. Only charge the auto-injector when the battery icon is flashing green or orange. Read more  pages 56–57.
pages 56–57.  pages 77-79. Storing the SKYTROFA Cartridge For information on the SKYTROFA Cartridge storing, see instructions on the cartridge pack and blister.
pages 77-79. Storing the SKYTROFA Cartridge For information on the SKYTROFA Cartridge storing, see instructions on the cartridge pack and blister. Product Information
| Page | |
|---|---|
| Product Safety | 70 |
| Expiration | 72 |
| EMC Compliance Levels | 74 |
| Technical Specification | 77 |
| Symbols | 80 |
| Warranty and Disclaimer | 84 |
| Parts Overview | 85 |
Product Safety
Do not use the SKYTROFA Auto-Injector if the SKYTROFA Cartridge cannot be inserted or removed, or if the cartridge was not completely emptied during the last attempted injection.
 | Do not use the auto-injector if you think it may be damaged. |
 | Do not open, try to repair, or change the auto-injector. The battery is not replaceable. Please call Ascendis Pharma Customer Support. For contact information |
 | Do not clean the auto-injector while charging. Keep the auto-injector dry. |
 | Do not use or place the auto-injector closer than 12 inches (30 cm) to microwave ovens or electronic equipment with antennas such as Wi-Fi transceivers. The auto-injector might not work the right way. |
 | Do not use a cartridge if it has been dropped or you think it may be damaged. |
 | Do not expose the auto-injector to extreme temperatures. Keep the auto-injector away from heat and open flames. Keep it out of direct sunlight. Do not place the auto-injector in liquid. Do not wash the auto-injector in a dishwasher. |
| Expiration | |
The SKYTROFA Auto-Injector has an expiration date (5 years after the manufacturing date) or a maximum of 210 injections, whichever comes first. The expiration date is located at the bottom of the auto-injector and is indicated by year (YYYY), month (MM), and date (DD), printed as YYYY-MM-DD. When there are 5 injections or less than 1 month left, the auto-injector will indicate this when turned on. Read more | |
 back cover.
back cover.  page 59. The auto-injector may be used for the remaining 5 injections. After the final injection or after the expiration date, your auto-injector has to be replaced. Please contact a healthcare provider or Ascendis Pharma Customer Support to receive a new auto-injector. For contact information
page 59. The auto-injector may be used for the remaining 5 injections. After the final injection or after the expiration date, your auto-injector has to be replaced. Please contact a healthcare provider or Ascendis Pharma Customer Support to receive a new auto-injector. For contact information  back cover.
back cover.  The auto-injector contains electrical and electronic components, including a non-replaceable battery. Do not throw away (dispose) in household trash. When the auto-injector reaches its expiration date, maximum number of injections, or needs replacing, return it to the Ascendis Pharma Customer Support. Contact Ascendis Pharma Customer Support for instructions on how to return properly. For contact information
The auto-injector contains electrical and electronic components, including a non-replaceable battery. Do not throw away (dispose) in household trash. When the auto-injector reaches its expiration date, maximum number of injections, or needs replacing, return it to the Ascendis Pharma Customer Support. Contact Ascendis Pharma Customer Support for instructions on how to return properly. For contact information  back cover.
back cover. EMC Compliance Levels
The auto-injector is intended for use in the electromagnetic environment specified below. The user of the auto-injector should ensure that it is used in such an environment.
| Electromagnetic Compatibility (EMC) Compliance Levels | Basic EMC Standard |
|---|---|
| EMC - RF Field radiated emission in 10 m (maximum): a) 30 MHz to 230 MHz: ≤ 30 dB (μV/m) quasi-peak. b) 230 MHz to 1 GHz: ≤ 37 dB (μV/m) quasi-peak. c) Internal source <108 MHz Compliance levels according to CISPR11, Group1, Class B. | IEC 60601-1-2 Edition 4.1/ EN 60601-1-2:2015/ A1:2021 CISPR 11 / EN 55011 EN ISO 11608- 4:2022 |
| EMC - RF Field radiated immunity - Electrical field: a) 10 V/m 80-2700 MHz. 1 kHz 80% AM | IEC 60601-1-2 Edition 4.1/ EN 60601-1-2:2015/ A1:2021 IEC 61000-4-3 EN ISO 11608- 4:2022 |
| EMC - Power Frequency Magnetic field, Immunity: a) 30 A/m | IEC 60601-1-2 Edition 4.1/ EN 60601-1-2:2015/ A1:2021 IEC 61000-4-3 EN ISO 11608- 4:2022 |
| EMC - Fast transients and bursts: a) Fast transients and bursts ±2 kV ac mains | IEC 60601-1-2 Edition 4.1/ EN 60601-1-2:2015/ A1:2021 IEC 61000-4-4 EN ISO 11608-4:2022 |
| EMC - Surge: a) Surge ±1 kV Line-to-Line ac mains | EC 60601-1-2 Edition 4.1/ EN 60601-1-2:2015/ A1:2021 IEC 61000-4-5 EN ISO 11608-4:2022 |
| EMC - Conducted immunity disturbance by RF fields a) 3 Vrms 150 kHz to 80 MHz (6 Vrms in ISM and amateur radio bands) | IEC 60601-1-2 Edition 4.1/ EN 60601-1-2:2015/ A1:2021 IEC 61000-4-6 EN ISO 11608-4:2022 |
| EMC - Conducted emission a) 150 kHz-30 MHz 120 V @ 60 Hz Compliance levels according to CISPR11, Group1, Class B. | IEC 60601-1-2 Edition 4.1 EN 60601-1-2:2015/ A1:2021 CISPR 11/EN 55011 EN ISO 11608-4:2022 |
| EMC - Voltage dips and interruptions: a) <5% UT for 5 sec b) <5% UT for 0.5 cycles c) 70% UT for 25 cycles d) 40% UT for 5 cycles | IEC 60601-1-2 Edition 4.1/ EN 60601-1-2:2015/ A1:2021 IEC 61000-4-11 EN ISO 11608-4:2022 |
| EMC - RF Field Immunity to Proximity fields from Wireless communication equipment, Immunity at 0.3 m separation: a) 27 V/m @ 380-390 MHz b) 28 V/m @ 430-470 MHz c) 9 V/m @ 704-787 MHz d) 28 V/m @ 800-960 MHz e) 28 V/m @ 1700-1990 MHz f) 28 V/m @ 2400-2570 MHz g) 9 V/m @ 5100-5800 MHz | IEC 60601-1-2 Edition 4.1/EN 60601-1-2:2015/ A1:2021 IEC 61000-4-39 EN ISO 11608- 4:2022 |
| EMC - Immunity to proximity magnetic fields (9 kHz to 13.56 MHz): a) 30 kHz - CW modulation - 8 A/m b) 134.2 kHz - 2.1 kHz pulse modulation - 65 A/m c) 13.56 MHz - 50 kHz pulse modulation - 7.5 A/m | IEC 60601-1-2 Edition 4.1/EN 60601-1-2:2015/ A1:2021 IEC 61000-4-39 EN ISO 11608- 4:2022 |
| ESD immunity: a) ± 8 kV contact b) ± 2, 4, 8 and 15 kV air | IEC 60601-1-2 Edition 4.1/EN 60601-1-2:2015/ A1:2021 IEC 61000-4-2 EN ISO 11608- 4:2022 |
Technical Specification
| SKYTROFA Auto-Injector | |
|---|---|
| Lifetime | Expiration date |
| Serial Number (SN) | Please refer to the bottom side of your auto-injector, marked after |
| Size | 7.09 × 1.46 × 1.06 in (180 × 37 × 27 mm). |
| Weight | 5.29 oz (150 g). |
| Display | Light-emitting diodes (LED). |
| Power source | Mains charger: 100 to 240 V AC. |
| Power supply | 1 rechargeable internal Li-ion battery. |
| Full capacity charging time | 2 hours and 30 minutes. Charging time for 1 injection: 15 minutes. |
| Full capacity | 3 weeks with 1 injection per week. |
| Electromagnetic compatibility | SKYTROFA Auto-Injector meets the requirements of IEC / EN 60601-1- 2, 4-th Edition. Using SKYTROFA Auto-Injector in the immediate vicinity of microwave appliances may result in impaired functioning. Use and store SKYTROFA Auto-Injector outside such an environment. |
| Protection against electric shock | Type BF. |
| Dose accuracy | SKYTROFA Auto-Injector meets the requirements of ISO 11608-1:2022. Minimum 90% of the volume. |
| Needle Insertion Depth | Based on needle length of 4 mm. |
| Ingress protection | IP22: IP2X: Ingress protection of solid foreign objects, ≥ 12.5mm diameter. IPX2: Ingress protection of water with harmful effects, dripping (15% tilted). Both put together called IP22. |
| Cable | USB to Micro-USB cable, 1m. |
| Bluetooth ® Version | BLE v5.3 with frequency 2.4 GHz. |
| Software Bill of Materials | The off-the-shelf software components that are used in the SKYTROFA Auto-Injector can be found on: https://www.phillipsmedisize. com/sbom |
 page 72.
page 72.  .
. | Charger | |
|---|---|
| Model | ASSA54a-050100. |
| Input | 100-240V – 50-60Hz. |
| Output | 5.0V |
 1.0A.
1.0A. | Use Conditions for SKYTROFA Auto-Injector | |
|---|---|
| Operation temperature (with medicine) and recommended storage temperature in between uses | 59°F to 86°F (15°C to 30°C) (marking on device bottom). |
| Storage & Transportation temperature | 14°F to 104°F (-10°C to 40°C) (marking on device cover). |
| Charging temperature | 59°F to 86°F (15°C to 30°C). |
| Operation humidity | 15 to 90% relative humidity, non-condensing. |
| Storage & Transportation relative humidity | Up to 93% relative humidity, non-condensing. |
| Atmospheric pressure | 10.15 psi to 15.37 psi (700 hPa to 1060 hPa). |
Use Conditions for Cartridge with Medicine and Needles
The use conditions provided here are for the SKYTROFA Auto-Injector device. The use conditions for SKYTROFA Cartridges with medicine and needles may be different. To see the use conditions for cartridges with medicine and needles, please see the information provided with your medicine.
Waste
To protect natural resources and to promote material reuse, please separate packaging material from electronic, medicine, and sharp waste and recycle the material through your local recycling system.
Electronic waste: The auto-injector, including battery, must be returned to the manufacturer. Contact Ascendis Pharma Customer Support for instructions on how to return properly  back cover.
back cover.
Charger and USB cable must be disposed of following local regulations for disposal of electronic waste.
Symbols
These symbols are found on the SKYTROFA Auto-Injector, the packaging, and the Instructions for Use.
 | Manufacturer. |
 | Distributor. |
 | Unique serial number of SKYTROFA Auto-Injector. |
 | Model Number. |
 | Manufacturer catalogue number for identification. |
 | Manufacturer batch code for identification. |
 | Medical Device. |
 | Single patient Multiple use. |
 | Expiration date. |
 | Consult Instructions For Use. |
 | Applied parts SKYTROFA Auto-Injector is a type BF device and provides protection against electrical shock and electrical current leakage. Applied parts on the device are the green top, the housing, and the button. |
 | Warning! When this warning symbol is seen, safety instructions must be followed. Otherwise, serious safety hazardous situations and injuries may occur. |
 | Precaution! When this precaution symbol is seen, safety instructions must be followed. Otherwise, minor to moderate safety hazardous situations and injuries and potential damage to the auto-injector or other property may occur. |
| Rx only | Caution: Federal law restricts this device for sale by or on the order of a physician. |
 | Operation temperature. |
 | Storage & Transportation temperature. |
 | Keep dry. |
 | If the auto-injector packaging has been damaged or unintentionally opened before the first use, contact customer support |
 | SKYTROFA Auto-Injector contains electrical and electronic components, including a battery that cannot be replaced, and must not be disposed of using standard waste collection. |
 | SKYTROFA Auto-injector packaging is a non-corrugated fiberboard. Can be disposed of as paper. |
 | SKYTROFA Auto-Injector has been tested and found to comply with the limits for a Class B digital device, according to part 15 of the FCC Rules. |
 | Radio transmitter – Bluetooth ® technology @2.4GHz using customized encrypted protocol – ClassB digital device. |
 | DC (Direct Current). |
 | Bluetooth ® technology |
 | Pairing key. |
 back cover.
back cover.
| The Unique Device Identifier (UDI) that appears on the carton is shown by a barcode and human readable format: (01) Device Identifier (10) Batch Number (17) Expiry date in YYMMDD format (21) Serial Number |

FCC Class B Notice
It is not allowed to modify the SKYTROFA Auto- Injector. Modifications may void the authority granted to the user by the FCC to operate this equipment.
This device complies with Part 15 of the FCC Rules. Operation is subject to the following two conditions:
- This device may not cause harmful interference.
- This device must accept any interference received, including interference that may cause undesired operation.
Note: This equipment has been tested and found to comply with the limits for a Class B digital device, pursuant to Part 15 of the FCC Rules.
These limits are designed to provide reasonable protection against harmful interference in a residential installation. This equipment generates, uses and can radiate radio frequency energy and, if not installed and used in accordance with the instructions, may cause harmful interference to radio communications. However, there is no guarantee that interference will not occur in a particular installation. If this equipment does cause harmful interference to radio or television reception, which can be determined by turning the equipment off and on, the user is encouraged to try to correct the interference by one or more of the following measures:
- Reorient or relocate the receiving antenna.
- Increase the separation between the equipment and receiver.
- Connect the equipment into an outlet on a circuit different from that to which the receiver is connected.
- Consult the dealer or an experienced radio/television technician for help.
Warranty and Disclaimer
Warranty
After the expiration date, your SKYTROFA Auto-Injector must be replaced. Please contact a healthcare provider or Ascendis Pharma Customer Support to receive a new auto-injector. For contact information  back cover.
back cover.
The expiration date is located at the bottom of the auto-injector and is indicated by year (YYYY), month (MM), and date (DD), printed as YYYY-MM-DD.
Disclaimer
The correct application of SKYTROFA according to the information provided with your medicine remains your responsibility.
Ascendis Pharma Endocrinology Inc. shall have no liability for incidental or consequential damages.
We reserve the right to modify technical specifications and documentation.
Patent Information
www.ascendispharma.us/products/patents
Parts Overview

Mechanism of Action
SKYTROFA is a pegylated human growth hormone (somatropin) for once-weekly subcutaneous injection [see Clinical Pharmacology (12.3) ] .
Somatropin binds to the growth hormone (GH) receptor in the cell membrane of target cells resulting in intracellular signal transduction and a host of pharmacodynamic effects. Somatropin has direct tissue and metabolic effects, and indirect effects mediated by insulin-like growth factor-1 (IGF-1), including stimulation of chondrocyte differentiation and proliferation, stimulation of hepatic glucose output, protein synthesis and lipolysis. Somatropin stimulates skeletal growth in pediatric patients with growth hormone deficiency (GHD) as a result of effects on the growth plates (epiphyses) of long bones.