Get your patient on Symproic (Naldemedine)
Patient education
Patient education materials
Dosing resources
Clinical information
Insurance resources
Prior authorization & coverage support
Financial assistance & copay programs
Other resources
Dosage & administration

Symproic prescribing information
| Warnings and Precautions, Gastrointestinal Perforation (5.1 ) | 07/2025 |
INDICATIONS AND USAGE
SYMPROIC is indicated for the treatment of opioid-induced constipation (OIC) in adult patients with chronic non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent (e.g., weekly) opioid dosage escalation.
DOSAGE AND ADMINISTRATION
Administration (2.1 ) :
- Alteration of analgesic dosing regimen prior to initiating SYMPROIC is not required
- Patients receiving opioids for less than 4 weeks may be less responsive to SYMPROIC
- Discontinue SYMPROIC if treatment with the opioid pain medication is also discontinued
Dosage (2.2 ) :
- In adults, the recommended dosage is 0.2 mg once daily with or without food
Administration
- Alteration of analgesic dosing regimen prior to initiating SYMPROIC is not required.
- Patients receiving opioids for less than 4 weeks may be less responsive to SYMPROIC [see Clinical Studies (14) ] .
- Discontinue SYMPROIC if treatment with the opioid pain medication is also discontinued.
Adult Dosage
The recommended dosage of SYMPROIC is 0.2 mg orally once daily with or without food.
DOSAGE FORMS AND STRENGTHS
Tablets: 0.2 mg naldemedine; supplied as yellow, round, film-coated, debossed with Shionogi marking above the identifier code 222 on one side and 0.2 on the other side.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
There are no available data with naldemedine in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. There is a potential for opioid withdrawal in a fetus when SYMPROIC is used in pregnant women [see Clinical Considerations ]. SYMPROIC should be used during pregnancy only if the potential benefit justifies the potential risk.
In a rat embryo-fetal development study following oral administration of naldemedine during the period of organogenesis at doses resulting in systemic exposure approximately 23,000 times the human area under the plasma-concentration time curve (AUC) at the recommended human dose of 0.2 mg/day, no developmental abnormalities were observed. In rabbits, there were no adverse effects on embryo-fetal development following oral administration of naldemedine during the period of organogenesis at doses resulting in systemic exposure approximately 226 times the human AUC at the recommended human dose of 0.2 mg/day [see Data ]. No effects on pre- and postnatal development were observed in rats at exposures 12 times human exposures at the recommended human dose.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Naldemedine crosses the placenta, and may precipitate opioid withdrawal in a fetus due to the immature fetal blood-brain barrier.
Data
Animal Data
In rats, there were no adverse effects on embryo-fetal development following oral administration of naldemedine during the period of organogenesis at doses up to 1000 mg/kg/day (approximately 23,000 times the human exposures (AUC) at the recommended human dose). In rabbits, there were no adverse effects on embryo-fetal development following oral administration of naldemedine during the period of organogenesis at doses up to 100 mg/kg/day (approximately 226 times the human exposures (AUC) at the recommended human dose). At 400 mg/kg/day (approximately 844 times the human exposures (AUC) at the recommended human dose), effects in maternal animals included body weight loss/decreased body weight gain and food consumption, fetal loss, and premature delivery. Decreased fetal body weights at this dose may be related to the maternal toxicity observed.
In the pre- and postnatal development study, pregnant rats were administered naldemedine at oral doses up to 1000 mg/kg/day from gestation day 7 through lactation day 20. No effects on pre- and postnatal development were observed in rats at 1 mg/kg/day (approximately 12 times the human exposures (AUC) at the recommended human dose). A single dam died at parturition at 1000 mg/kg/day, and decreased body weights/body weight gain and food consumption, poor nursing, and total litter loss were noted at 30 and 1000 mg/kg/day (approximately 626 and 17,000 times the human exposures (AUC) at the recommended human dose, respectively). Decreases in the offspring viability index on Day 4 after birth were noted at 30 and 1000 mg/kg/day, and low body weights and delayed pinna unfolding in pups were noted at 1000 mg/kg/day.
Lactation
Risk Summary
There is no information regarding the presence of naldemedine in human milk, the effects on the breastfed infant, or the effects on milk production. Naldemedine was present in the milk of rats [see Data ] . Because of the potential for serious adverse reactions, including opioid withdrawal in breastfed infants, a decision should be made to discontinue breastfeeding or discontinue the drug, taking into account the importance of the drug to the mother. If drug is discontinued in order to minimize drug exposure to a breastfed infant, advise women that breastfeeding may be resumed 3 days after the final dose of SYMPROIC.
Data
Drug-related radioactivity was transferred into milk of lactating rats following a single oral dose of 1 mg/kg [carbonyl- 14 C]-naldemedine.
Pediatric Use
The safety and effectiveness of SYMPROIC have not been established in pediatric patients.
Geriatric Use
Of the 1163 patients exposed to SYMPROIC in clinical studies, 183 (16%) were 65 years of age and over, while 37 (3%) were 75 years and over. No overall differences in safety or effectiveness between these and younger patients were observed, but greater sensitivity of some older individuals cannot be ruled out. In a population pharmacokinetic analysis, no age-related alterations in the pharmacokinetics of naldemedine were observed [see Clinical Pharmacology (12.3) ].
CONTRAINDICATIONS
SYMPROIC is contraindicated in:
- Patients with known or suspected gastrointestinal obstruction and patients at increased risk of recurrent obstruction, due to the potential for gastrointestinal perforation [see Warnings and Precautions (5.1) ].
- Patients with a history of a hypersensitivity reaction to naldemedine. Reactions have included bronchospasm and rash [see Adverse Reactions (6.1) ] .
WARNINGS AND PRECAUTIONS
- Gastrointestinal perforation : Consider the overall risk benefit in patients with known or suspected lesions of the GI tract. Monitor for severe, persistent, or worsening abdominal pain; discontinue if development of symptoms (5.1 )
- Opioid withdrawal : Consider the overall risk benefit in patients with disruptions to the blood-brain barrier. Monitor symptoms of opioid withdrawal (5.2 )
Gastrointestinal Perforation
Cases of gastrointestinal (GI) perforation have been reported with use of another peripherally acting opioid antagonist, including SYMPROIC. Postmarketing cases of GI perforation, including fatal cases, were reported when SYMPROIC was used in patients at risk of GI perforation (e.g., GI cancer, past GI surgery, diverticulitis, chemotherapy/radiation). SYMPROIC is contraindicated in patients with known or suspected gastrointestinal obstruction or in patients at risk of recurrent obstruction. Take into account the overall risk-benefit profile when using SYMPROIC in patients with these conditions or other conditions which might result in impaired integrity of the gastrointestinal tract wall (e.g., Crohn's disease). Monitor for the development of severe, persistent, or worsening abdominal pain; discontinue SYMPROIC in patients who develop this symptom.
Opioid Withdrawal
Clusters of symptoms consistent with opioid withdrawal, including hyperhidrosis, chills, increased lacrimation, hot flush/flushing, pyrexia, sneezing, feeling cold, abdominal pain, diarrhea, nausea, and vomiting have occurred in patients treated with SYMPROIC [see Adverse Reactions (6.1) ] .
Patients having disruptions to the blood-brain barrier may be at increased risk for opioid withdrawal or reduced analgesia. Take into account the overall risk-benefit profile when using SYMPROIC in such patients. Monitor for symptoms of opioid withdrawal in such patients.
ADVERSE REACTIONS
Serious and important adverse reactions described elsewhere in labeling include:
- Gastrointestinal perforation [see Warnings and Precautions (5.1) ]
- Opioid withdrawal [see Warnings and Precautions (5.2) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to SYMPROIC in 1163 patients in clinical trials, including 487 patients with exposures greater than six months and 203 patients with exposures of 12 months.
The following safety data are derived from three double-blind, placebo-controlled trials in patients with OIC and chronic non-cancer pain: two 12-week studies (Studies 1 and 2) and one 52-week study (Study 3) [see Clinical Studies (14) ].
In Studies 1 and 2, patients on laxatives were required to discontinue their use prior to study enrollment. All patients were restricted to bisacodyl rescue treatment during the study. In Study 3, approximately 60% of patients in both treatment groups were on a laxative regimen at baseline; patients were allowed to continue using their laxative regimen throughout the study duration. The safety profile of SYMPROIC relative to placebo was similar regardless of laxative use.
Tables 1 and 2 list common adverse reactions occurring in at least 2% of patients receiving SYMPROIC and at an incidence greater than placebo. Table 1 shows pooled 12-week data from Studies 1 and 2. Table 2 shows 12-week data from Study 3.
| Adverse Reaction | SYMPROIC 0.2 mg once daily N=542 | Placebo N=546 |
|---|---|---|
| Abdominal pain Abdominal pain includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper, gastrointestinal pain. | 8% | 2% |
| Diarrhea | 7% | 2% |
| Nausea | 4% | 2% |
| Gastroenteritis | 2% | 1% |
| Adverse Reaction | SYMPROIC 0.2 mg once daily N=621 | Placebo N=619 |
|---|---|---|
| Abdominal pain Abdominal pain includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper. | 11% | 5% |
| Diarrhea | 7% | 3% |
| Nausea | 6% | 5% |
| Vomiting | 3% | 2% |
| Gastroenteritis | 3% | 1% |
Adverse reactions up to 12 months in Study 3 are similar to those listed in Tables 1 and 2 (diarrhea: 11% vs. 5%, abdominal pain: 8% vs. 3%, and nausea: 8% vs. 6% for SYMPROIC and placebo, respectively).
Opioid Withdrawal
In Studies 1, 2 and 3, adverse reactions consistent with opioid withdrawal were based on investigator assessment and adjudicated based upon the occurrence of at least 3 adverse reactions potentially related to opioid withdrawal with onset of a constellation of those symptoms occurring on the same day or within one day of each other.
Adverse reactions of possible opioid withdrawal could include non-gastrointestinal (GI) symptoms (e.g., hyperhidrosis, hot flush or flushing, chills, tremor, tachycardia, anxiety, agitation, yawning, rhinorrhea, increased lacrimation, sneezing, feeling cold, and pyrexia), GI symptoms (e.g., vomiting, diarrhea, or abdominal pain), or both GI and non-GI symptoms.
In pooled Studies 1 and 2, the incidence of adverse reactions of opioid withdrawal was 1% (8/542) for SYMPROIC and 1% (3/546) for placebo. In Study 3 (52-week data), the incidence was 3% (20/621) for SYMPROIC and 1% (9/619) for placebo. Most SYMPROIC treated subjects experienced nearly equal incidence of GI only or both GI and non-GI symptoms.
Less Common Adverse Reactions:
Two patients developed symptoms of hypersensitivity following a single dose of SYMPROIC. One patient reported bronchospasm and another rash.
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of SYMPROIC. Because reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Gastrointestinal disorders : Gastrointestinal perforation [see Warnings and Precautions (5.1) ].
DRUG INTERACTIONS
Table 3 includes drugs with clinically important drug interactions with SYMPROIC and instructions for preventing or managing the interaction.
| Strong CYP3A Inducers (e.g., rifampin, carbamazepine, phenytoin, St. John's Wort) | |
| Clinical Impact | Significant decrease in plasma naldemedine concentrations, which may reduce efficacy [see Clinical Pharmacology (12.3) ] |
| Intervention | Avoid use of SYMPROIC with strong CYP3A inducers. |
| Other Opioid Antagonists | |
| Clinical Impact | Potential for additive effect of opioid receptor antagonism and increased risk of opioid withdrawal. |
| Intervention | Avoid use of SYMPROIC with another opioid antagonist. |
| Moderate (e.g., fluconazole, atazanavir, aprepitant, diltiazem, erythromycin) and Strong (e.g., itraconazole, ketoconazole, clarithromycin, ritonavir, saquinavir) CYP3A Inhibitors | |
| Clinical Impact | Increase in plasma naldemedine concentrations [see Clinical Pharmacology (12.3) ] |
| Intervention | Monitor for potential naldemedine-related adverse reactions [see Adverse Reactions (6.1) ] . |
| P-glycoprotein (P-gp) Inhibitors (e.g., amiodarone, captopril, cyclosporine, quercetin, quinidine, verapamil) | |
| Clinical Impact | Increase in plasma naldemedine concentrations [see Clinical Pharmacology (12.3) ] |
| Intervention | Monitor for potential naldemedine-related adverse reactions [see Adverse Reactions (6.1) ] . |
DESCRIPTION
SYMPROIC (naldemedine), an opioid antagonist, contains naldemedine tosylate as the active ingredient.
The chemical name for naldemedine tosylate is: 17-(cyclopropylmethyl)-6,7-didehydro-4,5α-epoxy-3,6,14-trihydroxy-N-[2-(3-phenyl-1,2,4-oxadiazol-5-yl)propan-2-yl]morphinan-7-carboxamide 4-methylbenzenesulfonic acid.
The structural formula is:
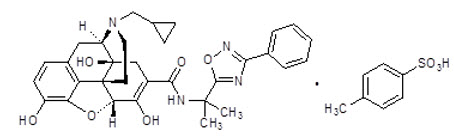
The empirical formula for naldemedine tosylate is C 32 H 34 N 4 O 6 ∙C 7 H 8 O 3 S and the molecular weight is 742.84.
Naldemedine tosylate is a white to light tan powder, soluble in dimethylsulfoxide and methanol, slightly soluble in alcohol and water, and independent of pH.
SYMPROIC (naldemedine) tablets for oral use contain 0.2 mg naldemedine (equivalent to 0.26 mg of naldemedine tosylate).
Excipients are: D-mannitol, croscarmellose sodium, magnesium stearate, hypromellose, talc, and yellow ferric oxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Naldemedine is an opioid antagonist with binding affinities for mu-, delta-, and kappa-opioid receptors. Naldemedine functions as a peripherally-acting mu-opioid receptor antagonist in tissues such as the gastrointestinal tract, thereby decreasing the constipating effects of opioids.
Naldemedine is a derivative of naltrexone to which a side chain has been added that increases the molecular weight and the polar surface area, thereby reducing its ability to cross the blood-brain barrier (BBB). Naldemedine is also a substrate of the P-glycoprotein (P-gp) efflux transporter. Based on these properties, the CNS penetration of naldemedine is expected to be negligible at the recommended dose levels, limiting the potential for interference with centrally-mediated opioid analgesia.
Pharmacodynamics
Use of opioids induces slowing of gastrointestinal motility and transit. Antagonism of gastrointestinal mu-opioid receptors by naldemedine inhibits opioid-induced delay of gastrointestinal transit time.
Effect on Cardiac Repolarization
At a dose up to 5 times the recommended dose, SYMPROIC does not prolong the QT interval to any clinically relevant extent.
Pharmacokinetics
Absorption
Following oral administration, naldemedine is absorbed with a time to achieve peak concentrations (T max ) of approximately 0.75 hours in a fasted state. Across the range of doses evaluated, the maximum plasma concentration (C max ) and area under the plasma concentration-time curve (AUC) increased in a dose-proportional or almost dose-proportional manner. Accumulation was minimal following multiple daily doses of naldemedine.
Food Effect
A high-fat meal decreased the rate but not the extent of naldemedine absorption. The C max was decreased by approximately 35% and time to achieve C max was delayed from 0.75 hours in the fasted state to 2.5 hours in the fed state, whereas there was no meaningful change in the AUC in the fed state [see Dosage and Administration (2.2) ] .
Distribution
Plasma protein binding of naldemedine in humans is 93% to 94%. The mean apparent volume of distribution during the terminal phase (Vz/F) is 155 L.
Elimination
The terminal elimination half-life of naldemedine is 11 hours.
Metabolism
Naldemedine is primarily metabolized by CYP3A to nor-naldemedine, with minor contribution from UGT1A3 to form naldemedine 3-G. Nor-naldemedine and naldemedine 3-G have been shown to have antagonistic activity for opioid receptors, with less potent effect than naldemedine.
Following oral administration of [ 14 C]-labeled naldemedine, the primary metabolite in plasma was nor-naldemedine, with a relative exposure compared to naldemedine of approximately 9% to 13%. Naldemedine 3-G was a minor metabolite in plasma, with a relative exposure to naldemedine of less than 3%.
Naldemedine also undergoes cleavage in the GI tract to form benzamidine and naldemedine carboxylic acid.
Excretion
Following oral administration of [ 14 C]-labeled naldemedine, the total amount of radioactivity excreted in the urine and feces was 57% and 35% of the administered dose of naldemedine, respectively. The amount of naldemedine excreted unchanged in the urine was approximately 16% to 18% of the administered dose. Benzamidine was the most predominant metabolite excreted in the urine and feces, representing approximately 32% and 20% of the administered dose of naldemedine, respectively. The percentage of unchanged drug in feces has not been estimated.
Use in Specific Populations
Age: Geriatric Population, Sex, Race/Ethnicity
A population pharmacokinetic analysis from clinical studies with naldemedine did not identify a clinically meaningful effect of age, sex, or race on the pharmacokinetics of naldemedine.
Renal Impairment
The pharmacokinetics of naldemedine after administration of a 0.2 mg single oral dose of SYMPROIC was studied in subjects with mild (n=8, estimated glomerular filtration rate [eGFR] of 60 to 89 mL/min/1.73 m 2 ), moderate (n=8, eGFR 30 to 59 mL/min/1.73 m 2 ), and severe (n=6, eGFR less than 30 mL/min/1.73 m 2 ) renal impairment, and subjects with end-stage renal disease (ESRD) requiring hemodialysis (n=8), and compared to healthy subjects with normal renal function (n=8, estimated creatinine clearance of at least 90 mL/min). The pharmacokinetics of naldemedine between subjects in all groups were similar.
Plasma concentrations of naldemedine in subjects with ESRD requiring hemodialysis were similar when SYMPROIC was administered either pre- or post-hemodialysis, indicating that naldemedine was not removed from the blood by hemodialysis.
Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of a 0.2 mg single oral dose of SYMPROIC was studied in subjects with hepatic impairment classified as mild (n=8, Child-Pugh Class A) or moderate (n=8, Child-Pugh Class B) and compared with healthy subjects with normal hepatic function (n=8). The pharmacokinetics of naldemedine between subjects in all groups were similar.
The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of naldemedine was not evaluated [see Use in Specific Populations (8.6) ] .
Drug Interaction Studies
Effect of Naldemedine on Other Drugs
In in vitro studies at clinically relevant concentrations, naldemedine did not inhibit the major CYP enzymes (including CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, or CYP4A11 isozymes) and is not an inhibitor of transporters (including OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, BCRP, or P-gp). Naldemedine did not cause significant induction of CYP1A2, CYP2B6, CYP3A4, UGT1A2, UGT1A6, or UGT2B7 isozymes.
Effect of Other Drugs on Naldemedine
Naldemedine is primarily metabolized by the CYP3A4 enzyme with a minor contribution from UGT1A3. Naldemedine is a substrate of P-gp. The effects of co-administered drugs on the pharmacokinetics of naldemedine are summarized in Figure 1.
Figure 1: Effect of Co-Administered Drugs on the Pharmacokinetics of Naldemedine
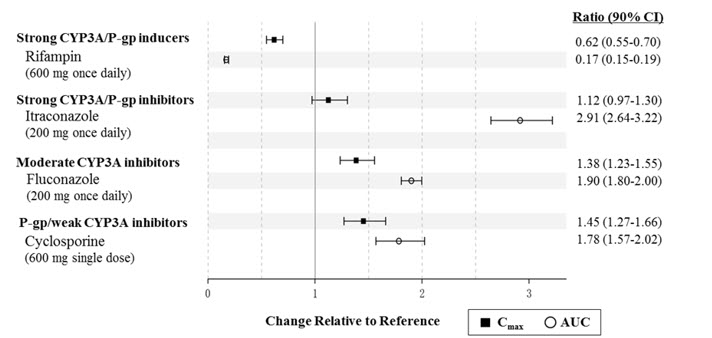
Efavirenz (moderate CYP3A inducer) : Simulation using physiologically-based pharmacokinetic modeling suggested that concomitant use of efavirenz decreases exposure to naldemedine by 43%. The clinical consequence of this decreased exposure is unknown.
No drug interaction studies have been conducted for SYMPROIC with drugs that alter gastric pH (e.g., antacids, proton-pump inhibitors).
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In 2-year carcinogenicity studies, there were no drug-related neoplastic findings following oral administration of naldemedine to mice and rats at doses up to 100 mg/kg/day (approximately 17,500 and 6,300 times the human exposures (AUC) at the recommended human dose, respectively).
Mutagenesis
Naldemedine was not genotoxic in the in vitro bacterial reverse mutation (Ames) assay, a chromosomal aberration assay with cultured Chinese hamster lung cells, or an in vivo micronucleus assay with rat bone marrow cells.
Impairment of Fertility
Naldemedine was found to have no effect on fertility or reproductive performance in male and female rats at oral doses up to 1000 mg/kg/day (approximately 17,000 times the human exposures (AUC) at the recommended human dose). In female rats, prolongation of diestrous phase was noted at 10 mg/kg/day (approximately 179 times the human exposures (AUC) at the recommended human dose).
CLINICAL STUDIES
SYMPROIC was evaluated in two replicate, 12-week, randomized, double-blind, placebo-controlled trials (Study 1 and Study 2) in which SYMPROIC was used without laxatives in patients with OIC and chronic non-cancer pain.
Patients receiving a stable opioid morphine equivalent daily dose of at least 30 mg for at least 4 weeks before enrollment and self-reported OIC were eligible for clinical trial participation.
Patients with evidence of significant structural abnormalities of the GI tract were not enrolled in these trials.
In Studies 1 and 2, patients had to either be not using laxatives or willing to discontinue laxative use at the time of screening and willing to use only the provided rescue laxatives during the screening and treatment periods.
In Studies 1 and 2, OIC was confirmed through a two-week run in period and was defined as no more than 4 spontaneous bowel movements (SBMs) total over 14 consecutive days and less than 3 SBMs in a given week with at least 25% of the SBMs associated with one or more of the following conditions: (1) straining; (2) hard or lumpy stools; (3) having a sensation of incomplete evacuation; and (4) having a sensation of anorectal obstruction/blockage.
An SBM was defined as a bowel movement (BM) without rescue laxative taken within the past 24 hours. Patients with no BMs over the 7 consecutive days prior to and during the 2-week screening period or patients who had never taken laxatives were excluded.
In the screening and treatment periods, bisacodyl was used as rescue laxative if patients had not had a BM for 72 hours and were allowed one-time use of an enema, if after 24 hours of taking bisacodyl they still had not had a BM.
A total of 547 patients in Study 1 and 553 patients in Study 2 were randomized in a 1:1 ratio to receive SYMPROIC 0.2 mg once daily or placebo for 12 weeks. Study medication was administered without regard to meals.
The mean age of subjects in Studies 1 and 2 was 54 years; 59% were women; and 80% were white. The most common types of pain in Studies 1 and 2 were back or neck pain (61%). The mean baseline number of SBMs was 1.3 and 1.2 per week for Studies 1 and 2, respectively.
Prior to enrollment, patients were using their current opioid for a mean duration of approximately 5 years. A wide range of types of opioids were used. The mean baseline opioid morphine equivalent daily dosage was 132 mg and 121 mg per day for Studies 1 and 2, respectively.
The efficacy of SYMPROIC was assessed in Studies 1 and 2 using a responder analysis. A responder was defined as a patient who had at least 3 SBMs per week and a change from baseline of at least 1 SBM per week for at least 9 out of the 12 weeks and 3 out of the last 4 weeks in Studies 1 and 2.
The responder rates in Studies 1 and 2 are shown in Table 4.
| Study 1 | Study 2 | |||||
|---|---|---|---|---|---|---|
| SYMPROIC 0.2 mg once daily (N=273) | Placebo (N=272) | Treatment Difference [95% CI] | SYMPROIC 0.2 mg once daily (N=276) | Placebo (N=274) | Treatment Difference [95% CI] | |
| CI=Confidence Interval | ||||||
| Responder The primary endpoint was defined as a patient who had at least 3 SBMs per week and a change from baseline of at least 1 SBM per week for at least 9 out of the 12 study weeks and 3 out of the last 4 weeks. | 130 (48%) | 94 (35%) | 13% [5%, 21%] | 145 (53%) | 92 (34%) | 19% [11%, 27%] |
| p value Cochran-Mantel-Haenszel test adjusted for opioid dose strata (30 to 100 mg; greater than 100 mg) | 0.0020 | <0.0001 | ||||
In Studies 1 and 2, the mean increase in frequency of SBMs per week from baseline to the last 2 weeks of the 12-week treatment period was 3.1 for SYMPROIC vs. 2.0 for placebo (difference 1.0, 95% CI 0.6, 1.5), and 3.3 for SYMPROIC vs. 2.1 for placebo (difference 1.2, 95% CI 0.8, 1.7), respectively.
During week 1 of the treatment period, the mean increase in frequency of SBMs per week from baseline was 3.3 for SYMPROIC vs. 1.3 for placebo (difference 2.0, 95% CI 1.5, 2.5) in Study 1 and 3.7 for SYMPROIC vs. 1.6 for placebo (difference 2.1, 95% CI 1.5, 2.6) in Study 2.
The mean increase in the frequency of complete SBM (CSBM) per week from baseline to the last 2 weeks of 12-week treatment period was 2.3 for SYMPROIC vs. 1.5 for placebo (difference 0.8, 95% CI 0.4, 1.2) in Study 1 and 2.6 for SYMPROIC vs. 1.6 for placebo (difference 1.1, 95% CI 0.6, 1.5) in Study 2. A CSBM was defined as a SBM that was associated with a sense of complete evacuation.
The change in the frequency of SBMs without straining per week from baseline to the last 2 weeks of the treatment period was 1.3 for SYMPROIC vs. 0.7 for placebo (difference 0.6, 95% CI 0.2, 0.9) in Study 1 and 1.8 for SYMPROIC vs. 1.1 for placebo (difference 0.7, 95% CI 0.3, 1.2) in Study 2.
HOW SUPPLIED/STORAGE AND HANDLING
SYMPROIC is supplied as 0.2 mg naldemedine tablets as follows:
- bottle of 30 tablets - NDC 59385-041-30
Store SYMPROIC in a light resistant container at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature].
Mechanism of Action
Naldemedine is an opioid antagonist with binding affinities for mu-, delta-, and kappa-opioid receptors. Naldemedine functions as a peripherally-acting mu-opioid receptor antagonist in tissues such as the gastrointestinal tract, thereby decreasing the constipating effects of opioids.
Naldemedine is a derivative of naltrexone to which a side chain has been added that increases the molecular weight and the polar surface area, thereby reducing its ability to cross the blood-brain barrier (BBB). Naldemedine is also a substrate of the P-glycoprotein (P-gp) efflux transporter. Based on these properties, the CNS penetration of naldemedine is expected to be negligible at the recommended dose levels, limiting the potential for interference with centrally-mediated opioid analgesia.