Get your patient on Trodelvy (Sacituzumab Govitecan)
Patient education
Administration guides
Patient education materials
Patient support program
Dosing resources
Clinical information
Insurance resources
Prior authorization & coverage support
Reimbursement information
Financial assistance & copay programs
Legal resources
Other resources
Dosage & administration

Trodelvy prescribing information
WARNING: NEUTROPENIA AND DIARRHEA
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm 3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia [see Dosage and Administration (2.4) ] . Initiate anti-infective treatment in patients with febrile neutropenia without delay [see Warnings and Precautions (5.1) ].
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide [see Warnings and Precautions (5.2) ]. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses [see Dosage and Administration (2.4) ].
INDICATIONS AND USAGE
TRODELVY is a Trop-2-directed antibody and topoisomerase inhibitor conjugate indicated:
Locally Advanced or Metastatic Triple-Negative Breast Cancer
First Line
- As a single agent for the first-line treatment of adult patients with unresectable locally advanced or metastatic triple negative breast cancer (TNBC) who are not candidates for PD-1 or PD-L1 inhibitor-based therapy. (1.1 , 14.1 )
- In combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph for the first-line treatment of adult patients with unresectable locally advanced or metastatic TNBC whose tumors express PD-L1 (CPS ≥ 10) as determined by an FDA-authorized test. (1.1 , 14.1 )
Second Line or Later
- For the treatment of adult patients with unresectable locally advanced or mTNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease. (1.1 , 14.1 )
Locally Advanced or Metastatic HR-Positive, HER2-Negative Breast Cancer
- For the treatment of adult patients with unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting. (1.1 , 14.2 )
Locally Advanced or Metastatic Triple-Negative Breast Cancer
First Line
- TRODELVY as a single agent is indicated for the first-line treatment of adult patients with unresectable locally advanced or metastatic triple negative breast cancer (TNBC) who are not candidates for PD-1 or PD-L1 inhibitor based therapy.
- TRODELVY, in combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph, is indicated for the first-line treatment of adult patients with unresectable locally advanced or metastatic TNBC whose tumors express PD-L1 [Combined Positive Score (CPS ≥ 10)] as determined by an FDA-authorized test [see Dosage and Administration (2.1) ] .
Second Line or Later
- TRODELVY is indicated for the treatment of adult patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
Locally Advanced or Metastatic HR-positive, HER2-negative Breast Cancer
- TRODELVY is indicated for the treatment of adult patients with unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
DOSAGE AND ADMINISTRATION
- Do NOT substitute TRODELVY for or use with other drugs containing irinotecan or its active metabolite SN-38. (2 )
- Premedication for prevention of infusion reactions and of chemotherapy-induced nausea and vomiting is recommended. (2.1 )
- The recommended dosage as a single agent or in combination with pembrolizumab is 10 mg/kg on Days 1 and 8 of 21-day cycles until disease progression or unacceptable toxicity. (2.3 )
- Monitor patients during the infusion and for at least 30 minutes after completion of infusion. Treatment interruption and/or dose reduction may be needed to manage adverse reactions. (2.4 , 2.5 )
- See Full Prescribing Information for preparation and administration instructions. (2.4 )
Important Use Information and Premedication
Do NOT substitute TRODELVY for or use with other drugs containing irinotecan or its active metabolite SN-38.
Premedication
Prior to each dose of TRODELVY, premedication for prevention of infusion reactions and of chemotherapy-induced nausea and vomiting (CINV) is recommended.
- Premedicate with antipyretics, H1 and H2 blockers prior to infusion, and corticosteroids may be used for patients who had prior infusion reactions.
- Premedicate with a 2 or 3 drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK 1 receptor antagonist, as well as other drugs as indicated).
Prophylaxis for Neutropenia
Primary prophylaxis with granulocyte colony-stimulating factor (G-CSF) is recommended starting in the first cycle for all patients at increased risk of febrile neutropenia [see Warnings and Precautions (5.1) ] .
Patient Selection for Combination Therapy
Select patients for treatment of unresectable locally advanced or metastatic TNBC with TRODELVY in combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph, based on the tumor expression of PD-L1 as confirmed by an FDA-authorized test [see Clinical Studies (14.1) ] .
Information on FDA-authorized tests is available at http://www.fda.gov/companiondiagnostics .
Recommended Dosage
The recommended dosage of TRODELVY as a single agent or in combination with pembrolizumab is 10 mg/kg administered as an intravenous infusion on Days 1 and 8 of each 21-day cycle. Continue TRODELVY until disease progression or unacceptable toxicity. Do not administer TRODELVY at doses greater than 10 mg/kg.
When TRODELVY is administered in combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph, discontinue pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph, after approximately 24 months.
Refer to the Prescribing Information for pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph for the recommended dosing information.
Dosage Modifications for Adverse Reactions
Management of adverse reactions may require temporary interruption, dose reduction, or permanent discontinuation of TRODELVY as described in Tables 1 and 2. Do not re-escalate the TRODELVY dose after a dose reduction for adverse reactions has been made.
| Dose Reduction Permanently discontinue TRODELVY in patients unable to tolerate 5 mg/kg. | Dosage and Schedule |
|---|---|
| First | Reduce to 7.5 mg/kg |
| Second | Reduce to 5 mg/kg |
The recommended dosage modifications for adverse reactions are provided in Table 2.
| Adverse reactions | Severity | Dose Modification |
|---|---|---|
| Neutropenia [see Warnings and Precautions (5.1) ] | Grade 3-4 neutropenia (Absolute Neutrophil Count [ANC] <1,000/mm 3 ) or febrile neutropenia |
|
| Nausea/Vomiting/ Diarrhea [see Warnings and Precautions (5.2 , 5.4) ] | Grade 3-4 nausea, vomiting or diarrhea that is not controlled with antiemetics or anti-diarrheal agents |
|
| Infusion-Related Reaction [see Warnings and Precautions (5.3) ] | Grade 1-3 infusion-related reactions |
|
| Grade 4 infusion-related reactions |
| |
| Other Toxicities | Other Grade 3-4 toxicities of any duration despite optimal medical management |
|
Dosage Modifications for Adverse Reactions for TRODELVY in Combination with Pembrolizumab or Pembrolizumab and berahyaluronidase alfa-pmph
Interrupt or discontinue one or both drugs of the combination or reduce the dose of TRODELVY to manage adverse reactions as appropriate. Refer to the prescribing information for pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph for recommendations on dosage interruption or discontinuation due to adverse reactions. For TRODELVY dosage modifications, refer to Table 1 and Table 2.
Preparation and Administration
Reconstitution
- TRODELVY is a hazardous drug. Follow applicable special handling and disposal procedures 1 .
- Calculate the required dose (mg) of TRODELVY based on the patient's current body weight [see Dosage and Administration (2.2) ] .
- Using a sterile syringe, slowly inject 20 mL of 0.9% Sodium Chloride Injection, USP, into each 180 mg TRODELVY vial. Each vial contains overfill to compensate for liquid loss during preparation and after reconstitution, the total resulting volume delivers a concentration of 10 mg/mL .
- Gently swirl vials and allow to dissolve for up to 15 minutes. Do not shake . Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The solution should be free of visible particulates, clear and yellow. Do not use the reconstituted solution if it is cloudy or discolored.
- Use reconstituted TRODELVY immediately to prepare a diluted TRODELVY infusion solution.
Dilution
- Calculate the required amount of the reconstituted TRODELVY solution needed to obtain the appropriate dose according to the patient's body weight.
- Determine the final volume of the infusion solution to deliver the appropriate dose at a TRODELVY concentration range of 1.1 mg/mL to 3.4 mg/mL.
- Use 0.9% Sodium Chloride Injection, USP only since the stability of the reconstituted TRODELVY solution has not been determined with other infusion-based solutions. Use a polyvinyl chloride, polypropylene/polyethylene, polyolefin, or ethylene vinyl acetate infusion bag.
- Withdraw and discard the volume of 0.9% Sodium Chloride Injection, USP from the final infusion bag that is necessary to achieve the indicated TRODELVY concentration following the addition of the calculated amount of reconstituted TRODELVY solution.
- Withdraw the calculated amount of the reconstituted TRODELVY solution from the vial(s) using a syringe. Discard any unused portion remaining in the vial(s).
- To minimize foaming, slowly inject the calculated amount of reconstituted TRODELVY solution into the infusion bag. Do not shake the contents.
- If not used immediately, the infusion bag containing TRODELVY solution can be stored refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours protected from light. After refrigeration, administer diluted solution at room temperature up to 25°C (77°F) within 8 hours (including infusion time). Do Not Freeze or Shake.
Administration
- Administer TRODELVY as an intravenous infusion.
- First infusion: Administer over 3 hours. Observe patients during the infusion and for at least 30 minutes following the initial dose, for signs or symptoms of infusion-related reactions [see Warning and Precautions (5.3) ] .
- Second and subsequent infusions : Administer over 1 to 2 hours if prior infusions were tolerated. Observe patients during the infusion and for at least 30 minutes after infusion.
- Protect infusion bag from light. The infusion bag should be covered during administration to the patient until dosing is complete. It is not necessary to cover the infusion tubing or to use light-protective tubing during the infusion.
- An infusion pump may be used.
- Do not mix TRODELVY, or administer in the same intravenous line, with other medicinal products.
- Upon completion of the infusion, flush the intravenous line with 20 mL 0.9% Sodium Chloride Injection, USP.
DOSAGE FORMS AND STRENGTHS
For injection: 180 mg off-white to yellowish lyophilized powder in a single-dose vial.
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. (8.2 )
Pregnancy
Risk Summary
Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. There are no available data in pregnant women to inform the drug-associated risk. TRODELVY contains a genotoxic component, SN-38, and is toxic to rapidly dividing cells [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1) ] . Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 – 4% and 15 – 20%, respectively.
Data
Animal data
There were no reproductive and developmental toxicology studies conducted with sacituzumab govitecan-hziy.
Lactation
Risk Summary
There is no information regarding the presence of sacituzumab govitecan-hziy or SN-38 in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment and for 1 month after the last dose of TRODELVY.
Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of TRODELVY.
Contraception
Females
TRODELVY can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1) ] . Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose.
Males
Because of the potential for genotoxicity, advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
Infertility
Females
Based on findings in animals, TRODELVY may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1) ] .
Pediatric Use
Safety and effectiveness of TRODELVY have not been established in pediatric patients.
Geriatric Use
As a Single Agent
Of the 641 patients with TNBC who were treated with TRODELVY in clinical studies, 20% of patients were 65 years and older and 5% were 75 years and older. No overall differences in effectiveness were observed between patients ≥ 65 years of age and younger adult patients. Patients 65 and older had an increased incidence of neutropenia with fatal outcomes.
Of the 322 patients with HR+/HER2- breast cancer who were treated with TRODELVY, 26% of patients were 65 years and older and 6% were 75 years and older. No overall differences in effectiveness were observed between patients ≥ 65 years of age and younger patients. There was a higher discontinuation rate due to adverse reactions in patients aged 65 years or older (14%) compared with younger patients (3%).
In Combination with Pembrolizumab
Of the 221 patients with TNBC who were treated with TRODELVY in combination with pembrolizumab in ASCENT-04, 26% of patients were 65 years and older and 5% were 75 years and older. No overall differences in effectiveness were observed between patients ≥ 65 years of age and younger adult patients. There was a higher rate of serious adverse reactions in patients aged 65 years or older (31%) compared with younger adult patients (26%).
Hepatic Impairment
The safety of TRODELVY in patients with moderate (total bilirubin > 1.5 to 3 × ULN) or severe (total bilirubin > 3 × ULN) hepatic impairment has not been established. TRODELVY has not been evaluated in patients with AST or ALT > 3 ULN without liver metastases, or AST or ALT > 5 ULN with liver metastases.
CONTRAINDICATIONS
TRODELVY is contraindicated in patients who have experienced a severe hypersensitivity reaction to TRODELVY [see Warnings and Precautions (5.3) ] .
WARNINGS AND PRECAUTIONS
- Hypersensitivity and Infusion-Related Reactions : Hypersensitivity reactions including severe anaphylactic reactions have been observed. Monitor patients for infusion-related reactions. Permanently discontinue TRODELVY if severe or life-threatening reactions occur. (5.3 )
- Nausea/Vomiting : Use antiemetic preventive treatment and withhold TRODELVY for patients with Grade 3 nausea or Grade 3-4 vomiting at the time of scheduled treatment. (5.4 )
- Patients with Reduced UGT1A1 Activity : Individuals who are homozygous for the UGT1A1•28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia following initiation of TRODELVY. (5.5 )
- Embryo-Fetal Toxicity : TRODELVY can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.6 , 8.1 , 8.3 )
Neutropenia
TRODELVY can cause severe, life-threatening, or fatal neutropenia as early as the first cycle of treatment. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 48% of patients. Febrile neutropenia occurred in 6% of patients. The median time to first onset of neutropenia (including febrile neutropenia) was 19 days (range: 1 to 1022 days). Neutropenia occurred earlier in patients with reduced UGT1A1 activity [see Warnings and Precautions (5.5) ] . Neutropenic colitis occurred in 1.4% of patients.
Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities [see Dosage and Administration (2.1) ] .
Monitor absolute neutrophil count (ANC) during treatment. Withheld TRODELVY for ANC below 1500/mm 3 on Day 1 of any cycle or below 1000/mm 3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Dose modifications may be required due to neutropenia. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 [see Dosage and Administration (2.4) ] .
Diarrhea
TRODELVY can cause severe diarrhea. Diarrhea occurred in 62% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 10% of all patients treated with TRODELVY. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.6% of all patients.
Withhold TRODELVY for Grade 3-4 diarrhea at the time of scheduled treatment administration and resume when resolved to ≤ Grade 1 [see Dosage and Administration (2.4) ].
At the onset of diarrhea, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte replacement) may also be employed as clinically indicated.
Patients who exhibit an excessive cholinergic response to treatment with TRODELVY (e.g., abdominal cramping, diarrhea, salivation, etc.) can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions
TRODELVY can cause serious hypersensitivity reactions including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, and skin reactions [see Contraindications (4) ] .
Hypersensitivity reactions occurred in 28% of patients treated with TRODELVY with 13% occurring within 24 hours of dosage. Grade 3-4 hypersensitivity occurred in 1.5% of patients treated with TRODELVY with 0.4% of these occurring within 24 hours of dosage. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. The incidence of anaphylactic reaction was <0.1%.
Premedication for infusion reactions in patients receiving TRODELVY is recommended . Have medications and emergency equipment to treat infusion-related reactions, including anaphylaxis, available for immediate use when administering TRODELVY [see Dosage and Administration (2.1) ].
Closely monitor patients for hypersensitivity and infusion-related reactions during each TRODELVY infusion and for at least 30 minutes after completion of each infusion [see Dosage and Administration (2.3) ] .
Permanently discontinue TRODELVY for Grade 4 infusion-related reactions [see Dosage and Administration (2.4) ] .
Nausea and Vomiting
TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 63% of all patients treated with TRODELVY. Grade 3-4 nausea occurred in 3% of patients.
Vomiting occurred in 33% of all patients treated with TRODELVY. Grade 3-4 vomiting occurred in 2% of these patients.
Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK 1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV) [ see Dosage and Administration (2.1) ].
Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting at the time of scheduled treatment administration and resume with additional supportive measures when resolved to ≤Grade 1 [see Dosage and Administration (2.4) ].
Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity
Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)•28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia; and may be at increased risk for other adverse reactions when treated with TRODELVY.
The incidence of neutropenia and anemia was analyzed in 1202 patients who received TRODELVY and had UGT1A1 genotype results. In patients homozygous for the UGT1A1 •28 allele (n=138), the incidence of Grade 3-4 neutropenia was 57%. In patients heterozygous for the UGT1A1•28 allele (n=531), the incidence of Grade 3-4 neutropenia was 48%. In patients homozygous for the wild-type allele (n=533), the incidence of Grade 3-4 neutropenia was 41% [see Clinical Pharmacology (12.5) ] . In patients homozygous for the UGT1A1 •28 allele, the incidence of Grade 3-4 anemia was 17%. In patients heterozygous for the UGT1A1•28 allele, the incidence of Grade 3-4 anemia was 9%. In patients homozygous for the wild-type allele, the incidence of Grade 3-4 anemia was 8%.
The median time to first neutropenia including febrile neutropenia was 9 days in patients homozygous for the UGT1A1•28 allele, 19 days in patients heterozygous for the UGT1A1•28 allele, and 21 days in patients homozygous for the wild-type allele. The median time to first anemia was 22 days in patients homozygous for the UGT1A1•28 allele, 29 days in patients heterozygous for the UGT1A1•28 allele, and 29 days in patients homozygous for the wild-type allele.
Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 enzyme activity [see Dosage and Administration (2.4) ].
Embryo-Fetal Toxicity
Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1) ]. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose [see Use in Specific Populations (8.1 , 8.3) ] .
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Neutropenia [see Warnings and Precautions (5.1) ]
- Diarrhea [see Warnings and Precautions (5.2) ]
- Hypersensitivity and Infusion-Related Reactions [see Warnings and Precautions (5.3) ]
- Nausea and Vomiting [see Warnings and Precautions (5.4) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The pooled safety population described in the Warnings and Precautions section reflect exposure to TRODELVY as a single agent in 1354 patients, which included 641 patients with mTNBC and 322 patients with hormone receptor-positive (HR+)/human epidermal growth factor receptor 2-negative (HER2-) breast cancer from ASCENT-03, IMMU-132-01, ASCENT, and TROPiCS-02; and 391 patients with other tumor types. TRODELVY was administered as an intravenous infusion once weekly on Days 1 and 8 of 21-day treatment cycles at doses of 10 mg/kg until disease progression or unacceptable toxicity. Among the 1354 patients treated with TRODELVY, the median duration of treatment was 4.9 months (range: 0 to 63 months). In this pooled safety population, the most common (> 25%) adverse reactions including laboratory abnormalities were decreased leukocyte count (83%), decreased neutrophil count (77%), decreased hemoglobin (71%), nausea (63%), diarrhea (62%), decreased lymphocyte count (60%), fatigue (59%), alopecia (47%), increased glucose (40%), constipation (37%), vomiting (33%), decreased albumin (32%), increased alkaline phosphatase (30%) decreased appetite (28%), abdominal pain (27%), decreased creatinine clearance (27%), decreased magnesium (26%), and decreased potassium (26%).
The data described in the following section reflects exposure to TRODELVY in combination with intravenous pembrolizumab in 221 patients with PD-L1 positive TNBC from ASCENT-04. Among the 221 patients who received TRODELVY in combination with intravenous pembrolizumab, the most common (≥ 25%) adverse reactions including laboratory abnormalities were decreased neutrophil count and decreased hemoglobin (86% each), decreased leukocyte count (84%), diarrhea (72%), nausea (68%), decreased lymphocyte count (61%), fatigue (58%), alopecia (52%), increased alkaline phosphatase and increased glucose (50% each), increased alanine aminotransferase (47%), constipation (41%), increased aspartate aminotransferase (40%), rash (37%), decreased potassium (35%), increased lactate dehydrogenase (34%), vomiting (29%), abdominal pain, headache, increased eosinophils (26% each) and decreased albumin (25%).
Locally Advanced or Metastatic Triple-Negative Breast Cancer (TNBC)
Single-Agent in Previously Untreated Unresectable Locally Advanced or Metastatic TNBC (ASCENT-03)
The safety of TRODELVY was evaluated in 275 patients with unresectable locally advanced or metastatic TNBC who had not received previous systemic therapy for advanced disease and who were not candidates for PD-1 or PD-L1 inhibitor therapy who had received at least one dose of TRODELVY 10 mg/kg in ASCENT-03. [see Clinical Studies (14.1) ] . The median duration of treatment was 8.3 months (range: 0 to 29 months).
Serious adverse reactions occurred in 26% of patients receiving TRODELVY. Serious adverse reactions in > 2% of patients included diarrhea, febrile neutropenia, and neutropenia (3.6% each) and pneumonia (2.9%). Fatal adverse reactions occurred in 2.5% of patients who received TRODELVY including sepsis (1.1%) and acute respiratory failure , neutropenic colitis, pneumonia, and septic shock (0.4% each).
Permanent discontinuation of TRODELVY due to adverse reactions occurred in 3.6% of patients, of which interstitial lung disease accounted for 1.1%.
Dosage interruptions of TRODELVY occurred in 66% of patients. Adverse reactions which required dosage interruption in ≥ 5% of patients were decreased neutrophil count (43%), diarrhea (6%), decreased leukocyte count and COVID-19 (5% each).
Dose reductions of TRODELVY due to an adverse reaction occurred in 37% of patients. Adverse reaction which required dose reductions in >2% of patients included decreased neutrophil count (18%), diarrhea (6%), fatigue (4.7%), febrile neutropenia (2.5%), and weight decreased (2.2%).
The most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased neutrophil count (84%), decreased leukocyte count (80%), decreased hemoglobin (78%), nausea (61%), diarrhea and alopecia (55% each), increased glucose (52%), decreased lymphocyte count and fatigue (47% each), increased alanine aminotransferase (39%), increased alkaline phosphatase and constipation (38% each), increased lactate dehydrogenase (35%), increased aspartate aminotransferase (31%), decreased potassium (28%) and vomiting (25%).
Tables 3 and 4 summarize the adverse reactions and laboratory abnormalities in ASCENT-03.
| TRODELVY (n=275) | Treatment of Physician’s Choice Treatment of Physician’s Choice included gemcitabine/carboplatin (n=122), nab-paclitaxel (n=110), and paclitaxel (n=44) (n=276) | |||
|---|---|---|---|---|
| Adverse Reaction Graded per NCI CTCAE v.5.0. | All Grades (%) | Grade 3 - 4 (%) | All Grades (%) | Grade 3 - 4 (%) |
| Gastrointestinal disorders | ||||
| Nausea | 61 | 1.8 | 34 | 0.4 |
| Diarrhea Includes other related terms | 55 | 10 | 20 | 0.7 |
| Constipation | 38 | 0 | 25 | 0 |
| Vomiting | 25 | 1.8 | 13 | 1.4 |
| Abdominal Pain | 24 | 0.7 | 12 | 0 |
| Stomatitis | 19 | 1.1 | 9 | 0.4 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 55 | 0 | 27 | 0 |
| Rash | 20 | 0.4 | 18 | 0.4 |
| Pruritus | 10 | 0 | 6 | 0 |
| General disorders and administration site conditions | ||||
| Fatigue | 47 | 3.3 | 47 | 4.0 |
| Edema | 11 | 0.4 | 11 | 0 |
| Pyrexia | 11 | 1.1 | 10 | 0.7 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Cough | 18 | 0 | 13 | 0.4 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 17 | 0.7 | 10 | 0.4 |
| Nervous system disorders | ||||
| Headache | 17 | 0.4 | 12 | 0 |
| Peripheral neuropathy | 12 | 0 | 31 | 0.4 |
| Infections and infestations | ||||
| Upper respiratory tract infection | 16 | 0.4 | 9 | 0 |
| Urinary tract infection | 10 | 0.7 | 15 | 0.7 |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia | 15 | 0 | 17 | 0.4 |
| Back pain | 11 | 0.4 | 8 | 0.4 |
| Laboratory Abnormality | TRODELVY (n=275) | Treatment of Physician’s Choice (n=276) | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 - 4 (%) | All Grades (%) | Grade 3 - 4 (%) | |
| Hematology | ||||
| Decreased neutrophil count | 84 | 47 | 80 | 43 |
| Decreased leukocyte count | 80 | 29 | 80 | 33 |
| Decreased hemoglobin | 78 | 7 | 81 | 19 |
| Decreased lymphocyte count | 47 | 16 | 53 | 19 |
| Decreased platelet count | 18 | 6 | 40 | 17 |
| Chemistry | ||||
| Increased glucose | 52 | 0 | 44 | 0 |
| Increased alanine aminotransferase | 39 | 4.0 | 56 | 6 |
| Increased alkaline phosphatase | 38 | 1.1 | 35 | 0 |
| Increased lactate dehydrogenase | 35 | 0 | 35 | 0 |
| Increased aspartate aminotransferase | 31 | 2.2 | 47 | 2.5 |
| Decreased potassium | 28 | 5 | 18 | 2.5 |
| Decreased albumin | 23 | 2.5 | 14 | 0.4 |
| Decreased magnesium | 19 | 2.9 | 25 | 0.7 |
| Decreased sodium | 18 | 1.5 | 15 | 1.1 |
| Increased phosphate | 16 | 0 | 9 | 0 |
| Increased potassium | 16 | 0.7 | 18 | 0.7 |
| Increased magnesium | 14 | 5 | 11 | 1.8 |
| Hypoglycemia | 14 | 1.1 | 9 | 0 |
| Decreased phosphate | 12 | 0 | 10 | 0 |
| Increased urate | 12 | 0 | 4 | 0 |
In Combination with Pembrolizumab in Previously Untreated, Unresectable Locally Advanced or Metastatic TNBC whose tumors express PD-L1 (ASCENT-04)
The safety of TRODELVY in combination with pembrolizumab was evaluated in 221 patients with unresectable locally advanced or metastatic TNBC who have not received previous systemic therapy for advanced disease and whose tumors express PD-L1 who received at least one dose of TRODELVY 10 mg/kg in combination with pembrolizumab. [see Clinical Studies (14.1) ] . The median duration of treatment of TRODELVY was 8.9 months (range: 0 to 27 months ).
Serious adverse reactions occurred in 38% of patients receiving TRODELVY in combination with pembrolizumab. Serious adverse reactions in ≥ 2% of patients included febrile neutropenia (7%), neutropenia (6%), diarrhea (5%), fatigue and pneumonia (2.3% each). Fatal adverse reactions occurred in 3.2% of patients who received TRODELVY in combination with pembrolizumab including death (unknown cause) (0.9%) and completed suicide, neutropenic sepsis, sepsis, pneumonia, and pulmonary embolism (0.5% each).
Permanent discontinuation of TRODELVY due to an adverse reaction occurred in 7% of patients. Adverse reactions which resulted in permanent discontinuation of TRODELVY in ≥1% of patients included infusion related reaction (0.9%).
Dosage interruptions of TRODELVY due to an adverse reaction occurred in 75% of patients. Adverse reactions which required dosage interruption in ≥5% of patients included neutropenia (44%), upper respiratory tract infection (10%), diarrhea (8%), COVID-19 (6%), and anemia and fatigue (5% each).
Dose reduction of TRODELVY due to an adverse reaction occurred in 35% of patients. Adverse reactions which required dose reductions in ≥5% of patients included neutropenia (15%), diarrhea (8%), and fatigue (6%).
G-CSF was used in 63% of patients who received TRODELVY in combination with pembrolizumab.
The most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased neutrophil count and decreased hemoglobin (86% each), decreased leukocyte count (84%), diarrhea (72%), nausea (68%), decreased lymphocyte count (61%), fatigue (58%), alopecia (52%), increased alkaline phosphatase and increased glucose (50% each), increased alanine aminotransferase (47%), constipation (41%), increased aspartate aminotransferase (40%), rash (37%), decreased potassium (35%), increased lactate dehydrogenase (34%), vomiting (29%), abdominal pain, headache and increased eosinophils (26% each), and decreased albumin (25%).
Tables 5 and 6 summarize the adverse reactions and laboratory abnormalities in ASCENT-04.
| TRODELVY plus pembrolizumab N=221 | Treatment of Physician’s Choice Treatment of Physician’s Choice included gemcitabine/carboplatin (n=107), nab-paclitaxel (n=68), and paclitaxel (n=45) plus pembrolizumab N=220 | |||
|---|---|---|---|---|
| Adverse Reactions Graded per NCI CTCAE v. 5.0 | All Grades (%) | Grade 3-4 (%) | All Grades (%) | Grade 3-4 (%) |
| Gastrointestinal disorders | ||||
| Diarrhea Includes other related terms | 72 | 12 | 30 | 2.7 |
| Nausea | 68 | 3.2 | 38 | 1.8 |
| Constipation | 41 | 0.5 | 35 | 0.5 |
| Vomiting | 29 | 0.9 | 14 | 1.8 |
| Abdominal Pain | 26 | 0.5 | 15 | 0 |
| Stomatitis | 18 | 0.5 | 16 | 0 |
| General disorders and administration site conditions | ||||
| Fatigue | 58 | 8 | 56 | 3.2 |
| Edema | 16 | 0.5 | 17 | 0.5 |
| Pyrexia | 12 | 0.9 | 12 | 0.5 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 52 | 0 | 32 | 0 |
| Rash | 37 | 1.4 | 33 | 1.8 |
| Pruritus | 14 | 0.5 | 12 | 0.9 |
| Nervous system disorders | ||||
| Headache | 26 | 0.5 | 18 | 0 |
| Peripheral neuropathy | 13 | 0.9 | 39 | 4.5 |
| Dizziness | 12 | 0 | 10 | 0 |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia | 19 | 0.9 | 24 | 0.5 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Cough | 19 | 0.5 | 20 | 0 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 18 | 1.8 | 14 | 0 |
| Hypothyroidism | 11 | 0.5 | 18 | 0 |
| Infections and infestations | ||||
| Upper respiratory tract infection | 18 | 0 | 13 | 0 |
| Urinary tract infection | 16 | 1.4 | 16 | 0.5 |
| COVID-19 | 10 | 0.5 | 7 | 0.5 |
| Reproductive system and breast disorders | ||||
| Breast pain | 10 | 0 | 9 | 0 |
| Investigations | ||||
| Weight decreased | 10 | 0 | 5 | 0.5 |
| Laboratory Abnormality | TRODELVY plus pembrolizumab N=221 | TPC plus pembrolizumab N=220 | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 - 4 (%) | All Grades (%) | Grade 3 - 4 (%) | |
| Hematology | ||||
| Decreased neutrophil count | 86 | 50 | 86 | 47 |
| Decreased hemoglobin | 86 | 10 | 88 | 18 |
| Decreased leukocytes count | 84 | 32 | 83 | 31 |
| Decreased lymphocyte count | 61 | 21 | 60 | 19 |
| Increased eosinophils | 26 | 0 | 17 | 0 |
| Decreased platelet count | 16 | 5 | 43 | 17 |
| Chemistry | ||||
| Increased alkaline phosphate | 50 | 0.9 | 33 | 1.8 |
| Increased glucose | 50 | 0 | 47 | 0 |
| Increased alanine aminotransferase | 47 | 4.1 | 55 | 8 |
| Increased aspartate aminotransferase | 40 | 3.7 | 51 | 4.1 |
| Decreased potassium | 35 | 4.6 | 24 | 2.3 |
| Increased lactate dehydrogenase | 34 | 0 | 37 | 0 |
| Decreased albumin | 25 | 0.9 | 14 | 0.9 |
| Decreased sodium | 20 | 1.4 | 20 | 3.2 |
| Increased Urate | 19 | 0 | 7 | 0 |
| Increased magnesium | 17 | 6 | 13 | 3.7 |
| Increased phosphate | 17 | 0 | 12 | 0 |
| Decreased magnesium | 16 | 1.4 | 21 | 0 |
| Increased thyroid stimulating hormone | 15 | 0 | 24 | 0 |
| Decreased phosphate | 14 | 0 | 9 | 0 |
| Increased blood bilirubin | 12 | 3.7 | 8 | 3.2 |
| Increased sodium | 12 | 0.5 | 2.3 | 0.5 |
| Decreased glucose | 11 | 0 | 15 | 1.4 |
| Increased potassium | 11 | 0.9 | 9 | 0.9 |
Previously Treated Locally Advanced or Metastatic TNBC (ASCENT)
The safety of TRODELVY was evaluated in 258 patients with metastatic TNBC who had previously received a taxane and at least two prior chemotherapies and who received at least one dose of TRODELVY 10 mg/kg in ASCENT. [see Clinical Studies (14.1) ] . The median duration of treatment was 4.4 months (range: 0 to 23 months).
Serious adverse reactions occurred in 27% of patients receiving TRODELVY. Serious adverse reactions in > 1% of patients receiving TRODELVY included neutropenia (7%), diarrhea (4%), and pneumonia (3%). Fatal adverse reactions occurred in 1.2% of patients who received TRODELVY, including respiratory failure (0.8%) and pneumonia (0.4%).
Permanent discontinuation of TRODELVY due to an adverse reaction occurred in 5% of patients. Adverse reactions which resulted in permanent discontinuation of TRODELVY in ≥1% of patients included pneumonia and fatigue (1% each).
Dosage interruptions of TRODELVY due to an adverse reaction occurred in 63% of patients. Adverse reactions which required dosage interruption in ≥5% of patients included neutropenia (47%), diarrhea (5%), respiratory infection (5%), and leukopenia (5%).
Dose reductions of TRODELVY due to an adverse reaction occurred in 22% of patients. Adverse reactions which required a dose reduction in >4% of patients included neutropenia (11%) and diarrhea (5%).
Granulocyte-colony stimulating factor (G-CSF) was used in 44% of patients who received TRODELVY.
The most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased hemoglobin (94%), decreased lymphocyte count (88%), decrease leukocyte count (86%), decreased neutrophil count (78%), fatigue (65%), diarrhea (59%), nausea (57%), increased glucose (49%), alopecia (47%), constipation (37%), decreased calcium (36%), vomiting, decreased magnesium, and decreased potassium (33% each), increased albumin (32%), abdominal pain (30%), decreased appetite (28%), increased aspartate aminotransferase (27%), increased alanine aminotransferase, increased alkaline phosphatase and decreased phosphate (26% each).
Tables 7 and 8 summarize adverse reactions and laboratory abnormalities, respectively, in ASCENT.
| TRODELVY (n=258) | Single Agent Chemotherapy Single agent chemotherapy included one of the following single-agents: eribulin (n=139), capecitabine (n=33), gemcitabine (n=38), or vinorelbine (except if patient had ≥ Grade 2 neuropathy, n=52). (n=224) | |||
|---|---|---|---|---|
| Adverse Reaction | All Grades % | Grade 3 - 4 % | All Grades % | Grade 3 - 4 % |
| i. Graded per NCI CTCAE v.5.0. | ||||
| General disorders and administration site conditions | ||||
| Fatigue Includes other related terms | 65 | 6 | 50 | 9 |
| Pyrexia | 15 | 0.4 | 14 | 2.2 |
| Gastrointestinal disorders | ||||
| Diarrhea | 59 | 11 | 17 | 0.9 |
| Nausea | 57 | 3.1 | 26 | 0.4 |
| Vomiting | 33 | 1.6 | 16 | 1.3 |
| Constipation | 37 | 0.4 | 23 | 0 |
| Abdominal Pain | 30 | 2.7 | 12 | 1.3 |
| Stomatitis | 17 | 1.6 | 13 | 1.3 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 47 | 0 | 16 | 0 |
| Rash | 12 | 0.4 | 5 | 0.4 |
| Pruritus | 10 | 0 | 3.1 | 0 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 28 | 1.6 | 21 | 0.9 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Cough | 24 | 0 | 18 | 0.4 |
| Nervous system disorders | ||||
| Headache | 18 | 0.8 | 13 | 0.4 |
| Dizziness | 10 | 0 | 7 | 0 |
| Musculoskeletal and connective tissue disorders | ||||
| Back pain | 16 | 1.2 | 14 | 1.8 |
| Arthralgia | 12 | 0.4 | 7 | 0 |
| Infections and infestations | ||||
| Urinary tract infection | 13 | 0.4 | 8 | 0.4 |
| Upper respiratory tract infection | 12 | 0 | 3.1 | 0 |
| Psychiatric disorders | ||||
| Insomnia | 11 | 0 | 5 | 0 |
| Laboratory Abnormality | TRODELVY (n=258) | Single Agent Chemotherapy (n=224) | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 - 4 (%) | All Grades (%) | Grade 3 - 4 (%) | |
| Hematology | ||||
| Decreased hemoglobin | 94 | 9 | 57 | 6 |
| Decreased lymphocyte count | 88 | 31 | 40 | 24 |
| Decreased leukocyte count | 86 | 41 | 53 | 25 |
| Decreased neutrophil count | 78 | 49 | 48 | 36 |
| Decreased platelet count | 23 | 1.2 | 25 | 2.7 |
| Chemistry | ||||
| Increased glucose | 49 | 2.3 | 43 | 2.8 |
| Decreased calcium | 36 | 1.6 | 21 | 1.4 |
| Decreased magnesium | 33 | 0.4 | 20 | 0 |
| Decreased potassium | 33 | 4.3 | 28 | 0.9 |
| Increased albumin | 32 | 0.8 | 25 | 1.4 |
| Increased aspartate aminotransferase | 27 | 1.2 | 32 | 1.4 |
| Increased alanine aminotransferase | 26 | 1.2 | 26 | 1.8 |
| Increased alkaline phosphatase | 26 | 0 | 17 | 0.5 |
| Decreased phosphate | 26 | 7.8 | 20 | 3.3 |
| Decreased sodium | 22 | 0.4 | 17 | 0.5 |
| Increased lactate dehydrogenase | 18 | 0 | 22 | 0 |
| Decreased glucose | 10 | 0 | 3.2 | 0 |
Study IMMU-132-01
The safety of TRODELVY was evaluated in 108 patients with metastatic TNBC who had received at least two prior anticancer therapies for metastatic disease who received at least one dose of TRODELVY at doses up to 10 mg/kg. [see Clinical Studies (14.1) ] . The median duration of treatment was 5.1 months (range: 0 to 51 months).
Serious adverse reactions occurred in 31% of patients receiving TRODELVY. Serious adverse reactions in > 1% of patients receiving TRODELVY included febrile neutropenia (6%) vomiting (5%), diarrhea (3.7%), nausea and dyspnea (2.8%), anemia, pleural effusion, neutropenia, pneumonia, dehydration (each 1.9%). A fatal adverse reaction of metastases to spine occurred in 1 patient (0.9%) who received TRODELVY.
Permanent discontinuation of TRODELVY due to an adverse reaction occurred in 2.8% of patients. Adverse reactions which resulted in permanent discontinuation of TRODELVY included anaphylaxis, decreased appetite, fatigue, and localized edema (each 0.9%).
Dosage interruptions of TRODELVY due to an adverse reaction occurred in 45% of patients. Adverse reactions which required dosage interruption in ≥2% of patients included neutropenia (33%), leukocytes decreased (6%), febrile neutropenia (3.7%), and anemia (2.8%).
Dose reductions of TRODELVY due to an adverse reaction occurred in 33% of patients. Adverse reactions which required dose reductions most commonly included neutropenia/febrile neutropenia.
The most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased hemoglobin (93%), decreased leukocyte count (91%), decreased neutrophil count (82%), nausea (69%), diarrhea (63%), increased activated partial thromboplastin time (60%), fatigue and increased alkaline phosphatase (57% each), vomiting and decreased calcium (49% each), increased aspartate aminotransferase (45%), decreased albumin (39%), alopecia (38%), increased alanine aminotransferase (35%), constipation (34%), rash and increased glucose (31% each), decreased appetite and decreased platelet count (30% each), decreased phosphate (29%), decreased magnesium (27%), abdominal pain (26%), respiratory infection (26%), and decreased sodium (25%).
Tables 9 and 10 summarize adverse reactions and laboratory abnormalities occurring in ≥10% of patients with metastatic TNBC in Study IMMU-132-01.
| Adverse Reaction | TRODELVY (n=108) | |
|---|---|---|
| All Grades (%) | Grade 3-4 (%) | |
| i. Graded per NCI CTCAE v. 4.0 | ||
| Any adverse reaction | 100 | 71 |
| Gastrointestinal disorders | 95 | 21 |
| Nausea | 69 | 6 |
| Diarrhea | 63 | 9 |
| Vomiting | 49 | 6 |
| Constipation | 34 | 0.9 |
| Abdominal pain Includes other related terms | 26 | 0.9 |
| Mucositis | 14 | 0.9 |
| General disorders and administration site conditions | 77 | 9 |
| Fatigue | 57 | 8 |
| Edema | 19 | 0 |
| Pyrexia | 14 | 0 |
| Metabolism and nutrition disorders | 68 | 22 |
| Decreased appetite | 30 | 0.9 |
| Dehydration | 13 | 5 |
| Skin and subcutaneous tissue disorders | 63 | 3.7 |
| Alopecia | 38 | 0 |
| Rash | 31 | 2.8 |
| Pruritus | 17 | 0 |
| Dry Skin | 15 | 0 |
| Nervous system disorders | 56 | 3.7 |
| Headache | 23 | 0.9 |
| Dizziness | 22 | 0 |
| Neuropathy | 24 | 0 |
| Dysgeusia | 11 | 0 |
| Infections and infestations | 55 | 12 |
| Respiratory Infection | 26 | 2.8 |
| Urinary Tract Infection | 21 | 2.8 |
| Musculoskeletal and connective tissue disorders | 54 | 0.9 |
| Back pain | 23 | 0 |
| Arthralgia | 17 | 0 |
| Pain in extremity | 11 | 0 |
| Respiratory, thoracic and mediastinal disorders | 54 | 5 |
| Cough | 22 | 0 |
| Dyspnea | 21 | 2.8 |
| Psychiatric disorders | 26 | 0.9 |
| Insomnia | 13 | 0 |
| Laboratory Abnormality | TRODELVY (n=108) | |
|---|---|---|
| All Grades (%) | Grade 3-4 (%) | |
| Hematology | ||
| Decreased hemoglobin | 93 | 6 |
| Decreased leukocyte count | 91 | 26 |
| Decreased neutrophil count | 82 | 32 |
| Increased activated partial thromboplastin time | 60 | 12 |
| Decreased platelet count | 30 | 2.8 |
| Chemistry | ||
| Increased alkaline phosphatase | 57 | 1.9 |
| Decreased calcium | 49 | 2.8 |
| Increased aspartate aminotransferase | 45 | 2.8 |
| Decreased albumin | 39 | 0.9 |
| Increased alanine aminotransferase | 35 | 1.9 |
| Increased glucose | 31 | 2.8 |
| Decreased phosphate | 29 | 5 |
| Decreased magnesium | 27 | 1.9 |
| Decreased sodium | 25 | 4.7 |
| Decreased potassium | 24 | 3.7 |
| Decreased glucose | 19 | 1.9 |
Locally Advanced or Metastatic HR-Positive, HER2-Negative Breast Cancer
TROPiCS-02
The safety of TRODELVY was evaluated in 268 patients with unresectable locally advanced or metastatic HR-positive, HER2-negative breast cancer whose disease has progressed after the following in any setting: a CDK 4/6 inhibitor, endocrine therapy, and a taxane; patients received at least two prior chemotherapies in the metastatic setting (one of which could be in the neoadjuvant or adjuvant setting if progression occurred within 12 months) who had received at least one dose of TRODELVY 10 mg/kg in TROPiCS-02. [see Clinical Studies (14.2) ] . The median duration of treatment was 4.1 months (range: 0 to 63 months).
Serious adverse reactions occurred in 28% of patients who received TRODELVY. Serious adverse reactions in >1% included diarrhea (5%), febrile neutropenia (4.1%), neutropenia (3.0%), abdominal pain (2.2%), neutropenic colitis , and vomiting (1.9 each%), and colitis and pneumonia 1.5% each). Fatal adverse reactions occurred in 2.2% of patients who received TRODELVY including arrhythmia, COVID-19 pneumonia, pneumonia, nervous system disorder, pulmonary embolism, and septic shock (0.4% each).
Permanent discontinuation of TRODELVY due to an adverse reaction occurred in 6% of patients. Adverse reactions which resulted in permanent discontinuation of TRODELVY in ≥ 0.5% of patients included asthenia, general physical health deterioration, and neutropenia (0.7% each).
Dosage interruptions of TRODELVY due to an adverse reaction occurred in 66% of patients. Adverse reactions which required dosage interruption in ≥ 5% of patients included neutropenia (50%).
Dose reductions of TRODELVY due to an adverse reaction occurred in 33% of patients. Adverse reactions which required dose reductions in >5% of patients included neutropenia (16%) and diarrhea (8%).
G-CSF was used in 54% of patients who received TRODELVY.
The most common (≥ 25%) adverse reactions, including laboratory abnormalities, were decreased leukocyte count (88%), decreased neutrophil count (83%), decreased hemoglobin (73%), decreased lymphocyte count (65%), diarrhea (62%), fatigue (60%), nausea (59%), alopecia (48%), increased glucose (37%), constipation (34%), and decreased albumin (32%).
Tables 11 and 12 summarize adverse reactions and laboratory abnormalities in TROPiCS-02.
| TRODELVY (n=268) | Single Agent Chemotherapy Single agent chemotherapy included one of the following single-agents: eribulin (n=130), vinorelbine (n=63), gemcitabine (n=56), or capecitabine (n=22). (n=249) | |||
|---|---|---|---|---|
| Adverse Reaction Graded per NCI CTCAE V 5.0 | All Grades % | Grade 3 - 4 % | All Grades % | Grade 3 - 4 % |
| Gastrointestinal disorders | ||||
| Diarrhea | 62 | 10 | 23 | 1.2 |
| Nausea | 59 | 1.1 | 35 | 2.8 |
| Constipation | 34 | 0.4 | 25 | 0 |
| Vomiting | 23 | 1.1 | 16 | 1.6 |
| Abdominal Pain | 20 | 3.7 | 14 | 0.8 |
| Dyspepsia Includes other related terms | 11 | 0 | 6 | 0 |
| General disorders and administration site conditions | ||||
| Fatigue | 60 | 8 | 51 | 4.4 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 48 | 0 | 19 | 0 |
| Pruritus | 12 | 0.4 | 2.4 | 0 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 21 | 1.5 | 21 | 0.8 |
| Hypokalemia | 10 | 1.5 | 3.6 | 0.4 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Dyspnea | 20 | 0 | 17 | 0 |
| Cough | 12 | 0 | 7 | 0.4 |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia | 15 | 0.7 | 12 | 0.4 |
| Nervous system disorders | ||||
| Headache | 16 | 0.4 | 15 | 0.4 |
Other clinically significant adverse reactions in TROPiCS-02 (≤ 10%) include: hypotension, pain, and rhinorrhea (4.9% each), hypocalcemia (3.0%), nasal congestion (2.6%), skin hyperpigmentation, (2.6%), colitis or neutropenic colitis (2.2%), pneumonia (1.9%), proteinuria (1.1%), enteritis (0.4%).
| Laboratory Abnormality | TRODELVY (n=268) | Single Agent Chemotherapy (n=249) | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3 - 4 (%) | All Grades (%) | Grade 3 - 4 (%) | |
| Hematology | ||||
| Decreased leukocyte count | 88 | 38 | 73 | 26 |
| Decreased neutrophil count | 83 | 53 | 67 | 40 |
| Decreased hemoglobin | 73 | 8 | 59 | 5 |
| Decreased lymphocyte count | 65 | 21 | 47 | 14 |
| Decreased platelet count | 21 | 1.1 | 30 | 3.7 |
| Eosinophilia | 13 | 0 | 4.1 | 0 |
| Chemistry | ||||
| Increased glucose | 37 | 0 | 31 | 0 |
| Decreased albumin | 32 | 0 | 27 | 0.4 |
| Decreased creatinine clearance | 24 | 2.3 | 19 | 1.3 |
| Increased alkaline phosphatase | 23 | 0 | 23 | 0.8 |
| Decreased potassium | 22 | 3.4 | 12 | 0.4 |
| Increased alanine aminotransferase | 21 | 1.1 | 31 | 2.1 |
| Decreased sodium | 19 | 0.8 | 17 | 0.4 |
| Decreased magnesium | 18 | 0 | 15 | 0 |
| Decreased phosphate | 17 | 0 | 10 | 0 |
| Increased phosphate | 16 | 0 | 16 | 0 |
| Increased lactate dehydrogenase | 16 | 0 | 28 | 0 |
| Increased aspartate aminotransferase | 15 | 1.5 | 25 | 1.3 |
| Increased potassium | 14 | 1.9 | 9 | 0 |
DRUG INTERACTIONS
- UGT1A1 Inhibitors or Inducers : Avoid concomitant use. (7 )
Effect of Other Drugs on TRODELVY
UGT1A1 Inhibitors
Avoid administering UGT1A1 inhibitors with TRODELVY.
SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38 [see Warnings and Precaution (5.5) and Clinical Pharmacology (12.3 , 12.5) ] .
UGT1A1 Inducers
Avoid administering UGT1A1 inducers with TRODELVY.
SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inducers of UGT1A1may reduce exposure to SN-38 [see Warnings and Precaution (5.5) and Clinical Pharmacology (12.3 , 12.5) ] .
DESCRIPTION
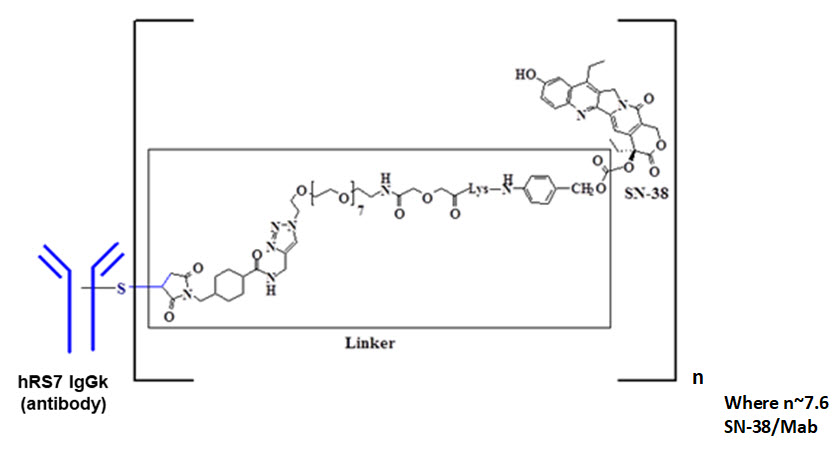
Sacituzumab govitecan-hziy is a Trop-2 directed antibody and topoisomerase inhibitor conjugate, composed of the following three components:
- the humanized monoclonal antibody, hRS7 IgG1κ (also called sacituzumab), which binds to Trop-2 (the trophoblast cell-surface antigen-2);
- the drug SN-38, a topoisomerase inhibitor;
- a hydrolysable linker (called CL2A), which links the humanized monoclonal antibody to SN-38.
The recombinant monoclonal antibody is produced by mammalian (murine myeloma) cells, while the small molecule components SN-38 and CL2A are produced by chemical synthesis. Sacituzumab govitecan-hziy contains on average 7 to 8 molecules of SN-38 per antibody molecule. Sacituzumab govitecan-hziy has a molecular weight of approximately 160 kilodaltons. Sacituzumab govitecan-hziy has the following chemical structure.
 |
TRODELVY (sacituzumab govitecan-hziy) for injection is a sterile, preservative-free, off-white to yellowish lyophilized powder for intravenous use in a 50 mL clear glass single-dose vial, with a rubber stopper and crimp-sealed with an aluminum flip-off cap.
Each single-dose vial of TRODELVY delivers 180 mg sacituzumab govitecan-hziy, 71.7 mg 2-(N-morpholino) ethane sulfonic acid (MES), 1.8 mg polysorbate 80 and 153.99 mg trehalose. Reconstitution with 20 mL of 0.9% Sodium Chloride Injection, USP, results in a concentration of 10 mg/mL with a pH of 6.5.
CLINICAL PHARMACOLOGY
Mechanism of Action
Sacituzumab govitecan-hziy is a Trop-2-directed antibody-drug conjugate. Sacituzumab is a humanized antibody that recognizes Trop-2. The small molecule, SN-38, is a topoisomerase I inhibitor, which is covalently attached to the antibody by a linker. Pharmacology data suggest that sacituzumab govitecan-hziy binds to Trop-2-expressing cancer cells and is internalized with the subsequent release of SN-38 via hydrolysis of the linker. SN-38 interacts with topoisomerase I and prevents re-ligation of topoisomerase I-induced single strand breaks. The resulting DNA damage leads to apoptosis and cell death. Sacituzumab govitecan-hziy decreased tumor growth in mouse xenograft models of triple-negative breast cancer.
Pharmacodynamics
The TRODELVY exposure-response relationships and pharmacodynamic time course for efficacy have not been fully characterized.
Cardiac electrophysiology
At the recommended dose, the maximum mean change from baseline was 9.7 msec (the upper bound of the two-sided 90% confidence interval is 16.8 msec). The increase in QTc was concentration dependent based on SN-38 concentrations.
Pharmacokinetics
The serum pharmacokinetics of sacituzumab govitecan-hziy and SN-38 were estimated in patients with mBC who received sacituzumab govitecan-hziy at the approved recommended dosage as a single agent or in combination with pembrolizumab and are presented as mean (%CV) unless otherwise specified. The pharmacokinetic parameters of sacituzumab govitecan-hziy and free SN-38 are presented in Table 13. No clinically significant differences in the pharmacokinetics of sacituzumab govitecan or SN-38 were observed when coadministered with pembrolizumab.
| Sacituzumab govitecan-hziy (N=827) | Free SN-38 (N=827) | |
|---|---|---|
| C max : maximum serum concentration from 0-168 hours after the first dose | ||
| AUC 0-168h : area under serum concentration curve through 168 hours after the first dose | ||
| C max [ng/mL] | 25,7107 (18%) | 108 (39%) |
| AUC 0-168h [ng•h/mL] | 12,049,500 (18%) | 3,510 (63%) |
Distribution
Sacituzumab govitecan-hziy steady state volume of distribution is 4.6 L.
Elimination
The median elimination half-life (t 1/2 ) of sacituzumab govitecan-hziy is 6.5 days and free SN-38 is 22 hours. The terminal half-life (t 1/2 ) of sacituzumab govitecan-hziy is 6.7 days (23%) and the apparent half-life (t 1/2 ) of free SN-38 is 24 hours (50%). The clearance of sacituzumab govitecan-hziy is 0.13 L/h (20%).
Renal elimination is known to contribute minimally to the excretion of SN-38, the small molecule moiety of sacituzumab govitecan-hziy.
Metabolism
No metabolism studies with sacituzumab govitecan-hziy were conducted. SN-38 (the small molecule moiety of sacituzumab govitecan-hziy) is metabolized via UGT1A1. The glucuronide metabolite of SN-38 (SN-38G) was detectable in the serum of patients.
Specific Populations
No clinically significant differences in the pharmacokinetics of sacituzumab govitecan-hziy were observed based on: age (27 to 88 years), race (75% White, 8% Asian, or 5% Black), or CLcr 30 to 89 mL/min. There are no data on the pharmacokinetics of sacituzumab govitecan-hziy in patients with CLcr 15 to 29 mL/min, or end-stage renal disease (CLcr < 15 mL/min).
Patients with Hepatic Impairment
The exposure of sacituzumab govitecan-hziy for patients with mild hepatic impairment (total bilirubin ≤ ULN with AST > ULN, or bilirubin > 1 to ≤ 1.5 ULN with any AST) is within range of that of patients with normal hepatic function (total bilirubin and AST < ULN).
Sacituzumab govitecan-hziy and free SN-38 exposures are unknown in patients with moderate (total bilirubin > 1.5 to 3 × ULN) or severe (total bilirubin > 3 × ULN) hepatic impairment.
Drug Interaction Studies
No drug-drug interaction studies were conducted with sacituzumab govitecan-hziy or its components. Inhibitors or inducers of UGT1A1 may increase or decrease SN-38 exposure, respectively.
Pharmacogenomics
SN-38 is metabolized via UGT1A1 [see Clinical Pharmacology (12.3) ] . Genetic variants of the UGT1A1 gene such as the UGT1A1•28 allele lead to reduced UGT1A1 enzyme activity. Individuals who are homozygous or heterozygous for the UGT1A1•28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia from TRODELVY compared to individuals who are wildtype (•1/•1) [see Warnings and Precautions (5.5) ].
Approximately 20% of the Black or African American population, 10% of the White population, and 2% of the East Asian population are homozygous for the UGT1A1•28 allele (•28/•28). Approximately 40% of the Black or African American population, 50% of the White population, and 25% of the East Asian population are heterozygous for the UGT1A1•28 allele (•1/•28). Decreased function alleles other than UGT1A1•28 may be present in certain populations.
Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of sacituzumab govitecan-hziy or of other sacituzumab govitecan products.
Among patients who received TRODELVY as a single agent over a median 5-month treatment period across 5 clinical studies, 1.2% (13/1058) of patients developed treatment-emergent ADA to sacituzumab govitecan and 77% (10/13) of ADA-positive patients developed neutralizing antibodies against sacituzumab govitecan. Among patients who received TRODELVY in combination with intravenous pembrolizumab in ASCENT-04, no treatment-emergent ADA was observed in 207 patients who were evaluable for ADA incidence. Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of sacituzumab govitecan-hziy is unknown.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with sacituzumab govitecan-hziy.
SN-38 was clastogenic in an in vitro mammalian cell micronucleus test in Chinese hamster ovary cells and was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
Fertility studies with sacituzumab govitecan-hziy have not been conducted. In a repeat-dose toxicity study in cynomolgus monkeys, intravenous administration of sacituzumab govitecan-hziy on Day 1 and Day 4 resulted in endometrial atrophy, uterine hemorrhage, increased follicular atresia of the ovary, and atrophy of vaginal epithelial cells at doses ≥ 60 mg/kg ( ³ 6 times the human recommended dose of 10 mg/kg based on body weight).
CLINICAL STUDIES
Locally Advanced or Metastatic Triple-Negative Breast Cancer (TNBC)
Single Agent in Previously Untreated, Unresectable Locally Advanced or Metastatic TNBC
ASCENT-03
The efficacy of TRODELVY was evaluated in ASCENT-03 (NCT05382299), a multicenter, open-label, randomized study that enrolled 558 patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who had not received previous systemic therapy for advanced disease and who were not candidates for PD-1 or PD-L1 inhibitor therapy. Patients were enrolled if:
- Their tumors were PD-L1 negative (defined as having a tumor CPS < 10 using the IHC 22C3 assay), or
- Their tumors were PD-L1 positive (defined as having a CPS ≥ 10 using the IHC 22C3 assay) and they had received a PD-1 or PD-L1 inhibitor in the (neo)adjuvant setting or if they had a comorbidity precluding treatment with a PD-1 or PD-L1 inhibitor.
Patients were excluded if they had received anticancer treatment or surgery within the previous 6 months, had active central nervous system (CNS) metastases and ECOG performance status (PS) >1.
Randomization was stratified by de novo metastatic disease vs recurrent disease within 6 to 12 months from completion of treatment in the curative setting versus recurrent disease > 12 months from completion of treatment in the curative setting, and by geographic region (United States/Canada/Western Europe vs. Rest of World).
Patients were randomized (1:1) to one of the following treatment arms; all study medications were administered via intravenous infusion:
- TRODELVY 10 mg/kg on Days 1 and 8 of a 21-day cycle (n=279)
- nab-paclitaxel 100mg/m 3 on Days 1, 8, and 15 of a 28-day cycle (n=110), paclitaxel 90 mg/m 2 on days 1, 8, and 15 of a 28-day cycle (n=45), or gemcitabine 1000 mg/m 2 and carboplatin AUC2 on Days 1 and 8 of a 21-day cycle (n=124)
Assessment of tumor status was performed every 6 weeks for the first year followed by every 12 weeks thereafter. Crossover to TRODELVY monotherapy was allowed at the time of disease progression and study treatment discontinuation. The primary efficacy outcome was progression-free survival (PFS) as assessed by Blinded Independent Central Review (BICR) per Response Evaluation Criteria in Solid Tumors (RECIST v1.1). Additional efficacy outcomes measures included overall survival (OS) and objective response rate (ORR).
The median age was 55 years (range: 23-86 years), 26% age 65 or older; 99.5% female; 64% White, 23% Asian, 4.5% American Indian or Alaskan Native, 3.0% Black; and 5.2% not specified , 72% non-Hispanic/non-Latino, 27% Hispanic/Latino, and 0.9% not reported; and 66% ECOG PS of 0 and 34% ECOG PS of 1. Of patients enrolled 31% had de novo disease, 21% had recurrent disease with a disease-free interval (DFI) of 6 to 12 months and 48% had recurrent disease with a DFI of > 12 months. Seventy-six percent of patients had visceral metastasis at baseline; and 5% had previously treated brain metastases. The majority of patients (99.5%) had tumor CPS < 10, and 0.4% had tumor CPS ≥ 10.
The trial demonstrated a statistically significant improvement in PFS. The OS data were immature and a total of 283 (51%) patients had died across both study arms.
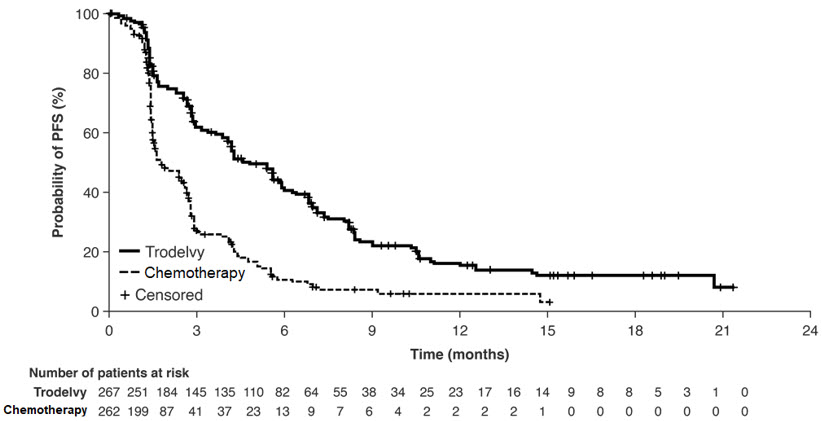
Table 14 and Figure 1 summarize the efficacy results for ASCENT-03.
| TRODELVY N=279 | TPC N=279 | |
|---|---|---|
| BICR = Blinded Independent Central Review; CI = Confidence Interval; TPC = Treatment of physician’s choice (gemcitabine and carboplatin, paclitaxel, or nab-paclitaxel) | ||
| Progression-Free Survival by BICR | ||
| Number of patients with events (%) | 161 (58) | 188 (67) |
| Median PFS in months (95% CI) | 9.7 (8.1, 11.1) | 6.9 (5.6, 8.2) |
| Hazard ratio (95% CI) Hazard ratio based on the stratified Cox proportional hazards model | 0.62 (0.50, 0.77) | |
| p-value 2-sided p-value based on stratified log-rank test | <0.0001 | |
| Objective Response Rate by BICR | ||
| Patients with Measurable Disease at Baseline, N | 266 | 264 |
| ORR (95% CI) | 50% (44, 56) | 47% (41, 53) |
| Complete response rate | 7% | 5% |
| Partial response rate | 43% | 42% |
Figure 1: Kaplan Meier Plot of Progression Free Survival (PFS) by BICR in ASCENT-03
In Combination with Pembrolizumab in Previously Untreated, Unresectable Locally Advanced or Metastatic TNBC whose tumors express PD-L1
ASCENT-04
The efficacy of TRODELVY in combination with intravenous pembrolizumab was evaluated in ASCENT-04 (NCT5382286) a multicenter, open-label, randomized study that enrolled 443 patients with locally advanced or metastatic TNBC who had not received previous systemic therapy for advanced disease and whose tumors express PD-L1 (CPS ≥ 10)] according to the PD-L1 IHC 22C3 pharmDx assay. Patients may have received chemotherapy with or without a PD-1 or PD-L1 inhibitor and/or radiotherapy in early-stage TNBC: however, at least 6 months must have elapsed between the completion of systemic breast cancer therapy or surgery, and first local or distant recurrence. Patients with active autoimmune disease that required systemic therapy within 2 years or treatment or a medical condition that required immunosuppression were ineligible.
Randomization was stratified by de novo metastatic disease versus disease recurrence within 6 to 12 months from completion of treatment in the curative setting versus disease recurrence > 12 months from completion of treatment in the curative setting, by geographic region (United States/ Canada/ Western Europe versus rest of world), and prior exposure to PD-1 or PD-L1 inhibitor (yes versus no).
Patients were randomized (1:1) to receive one of the following treatment arms; all study medications were administered via intravenous infusion:
- TRODELVY 10 mg/kg on Day 1 and 8 of 21-day cycles in combination with pembrolizumab 200 mg on Day 1 of 21-day cycles (n=221)
- nab-paclitaxel 100 mg/m 2 on Days 1, 8 and 15 every 28 days (n=69), paclitaxel 90 mg/m 2 on Days 1, 8, and 15 every 28 days (n=45), or gemcitabine 1000 mg/m 2 and carboplatin AUC 2 mg/mL/min on Days 1 and 8 every 21 days in combination with pembrolizumab 200 mg on Day 1 of 21-day cycles (n=108)
Assessment of tumor status was performed every 8 weeks for the first 18 months followed by every 12 weeks thereafter. Treatment beyond BICR-verified PD per RECIST was permitted if the patient was clinically stable and there was evidence of clinical benefit per the investigator. Crossover to TRODELVY monotherapy was allowed following disease progression and study treatment discontinuation. The primary efficacy outcome was progression-free survival (PFS) by BICR per RECIST v1.1. Additional efficacy outcomes measures included overall survival (OS) and objective response rate (ORR).
The median age was 55 years (range: 23-88 years); 26% age 65 years or older; 100% female; 58% White, 24% Asian, 6% American Indian or Alaska Native, 5% Black, and 6.5% not specified ; 29% Hispanic/Latino, 69% non-Hispanic/non-Latino and 2.0% not reported; and 70% ECOG PS of 0, 30% ECOG PS of 1, and 0.2% ECOG PS of 2. Of the patients enrolled 34% had de novo metastatic disease, 18% had recurrent disease with a DFI of 6 to 12 months and 48% had recurrent DFI of > 12 months. Sixty-five percent of patients had visceral metastasis at baseline; and 3% of patients had previously treated brain metastasis.
The trial demonstrated a statistically significant improvement in PFS. The OS data were immature and a total of 203 (46%) patients had died across both study arms.
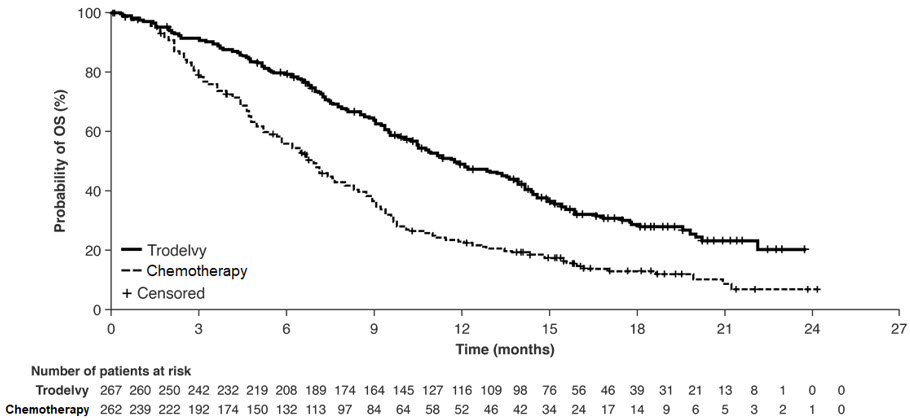
Table 15 and Figure 2 summarize the efficacy results for ASCENT-04.
| TRODELVY plus pembrolizumab N=221 | TPC plus pembrolizumab N=222 | |
|---|---|---|
| BICR = Blinded Independent Central Review; CI = Confidence Interval; TPC=Treatment of Physicians choice (nab-paclitaxel, paclitaxel, or gemcitabine with carboplatin) | ||
| Progression-Free Survival by BICR | ||
| Number of patients with events (%) | 109 (49) | 140 (63) |
| Median PFS in months (95% CI) | 11.2 (9.3, 16.7) | 7.8 (7.3, 9.3) |
| Hazard ratio (95% CI) Hazard ratio based on the stratified Cox proportional hazards model | 0.65 (0.51, 0.84) | |
| p-value 2-sided p-value based on stratified log-rank test | 0.0009 | |
| Objective Response Rate by BICR | ||
| Patients with Measurable Disease at Baseline, N | 210 | 213 |
| ORR (95% CI) | 61% (55, 68) | 55% (48, 62) |
| Complete response rate | 12% | 8% |
| Partial response rate | 50% | 47% |
Figure 2: Kaplan Meier Plot of Progression Free Survival (PFS) by BICR in ASCENT-04
Previously Treated, Locally Advanced or Metastatic TNBC
ASCENT
The efficacy of TRODELVY was evaluated in ASCENT (NCT02574455), a multicenter, open-label, randomized study conducted in 529 patients with unresectable locally advanced or metastatic TNBC who were previously treated with at least two prior lines of chemotherapy, one of which could be in the neoadjuvant setting provided progression occurred within a 12-month period. Patients were required to have received treatment with a taxane (unless contraindicated or not tolerated) in the neoadjuvant, adjuvant, or advanced setting. Patients with brain metastases were eligible to enroll up to a pre-defined maximum of 15% of patients. Magnetic resonance imaging (MRI) to determine brain metastases was required prior to enrollment for patients with known or suspected brain metastases. Patients with known Gilbert’s disease or bone-only disease were excluded.
Randomization was stratified by the number of prior chemotherapies (2-3 vs > 3), geographic region (North America vs Europe), and presence of brain metastasis (yes vs no).
Patients were randomized (1:1) to one of the following treatment arms:
- TRODELVY 10 mg/kg via intravenous infusion on Days 1 and 8 of a 21-day cycle (n=267)
- eribulin 1.23 or 1.4 mg/m 2 via intravenous infusion on Days 1 and 8 of a 21-day cycle (n=139), capecitabine 1000-1250 mg/m 2 orally twice daily on days 1-14 of a 21-day cycle (n=33), gemcitabine 800-1200 mg/m 2 via intravenous infusion on Days 1, 8, and 15 of a 28 day cycle (n=38), or vinorelbine 25 mg/m 2 via intravenous infusion on Day 1 weekly (n=52).
Assessment of tumor status was performed every 6 weeks for 36 weeks, then every 9 weeks thereafter. The primary efficacy outcome was progression-free survival (PFS) in patients without brain metastases at baseline (BICR per RECIST v1.1). Additional efficacy outcome measures included PFS for the full population and overall survival (OS).
The median age was 54 years (range: 27 to 82 years); 19% age 65 or older; 99.6% female; 79% White, 12% Black, 4.2% Asian and 5% not specified; 43% ECOG PS 0 and 57% ECOG PS 1; and 8.1% with BRCA1/BRCA2 mutations. Forty-two percent of patients had hepatic metastases, and 12 had previously treated, stable brain metastases. Twenty-nine percent received prior PD-1/PD-L1 therapy and 13% in the TRODELVY arm received only 1 prior line of systemic therapy in the metastatic setting.
The study demonstrated a statistically significant PFS. Efficacy results for the subgroup of patients who had received only 1 prior line of systemic therapy in the metastatic setting (in addition to having disease recurrence or progression within 12 months of neoadjuvant/adjuvant systemic therapy) were consistent with those who had received at least two prior lines in the metastatic setting.
Table 16, Figure 3 and Figure 4 summarize the efficacy results for ASCENT.
| All Randomized Patients | ||
|---|---|---|
| TRODELVY n=267 | Single Agent Chemotherapy n=262 | |
| CI = Confidence Interval | ||
| Progression-Free Survival PFS is defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever comes first. per BICR | ||
| Disease Progression or Death (%) | 190 (71%) | 171 (65%) |
| Median PFS in months (95% CI) | 4.8 (4.1, 5.8) | 1.7 (1.5, 2.5) |
| Hazard ratio Stratified log-rank test adjusted for stratification factors: number of prior chemotherapies, presence of known brain metastases at study entry, and region. (95% CI) | 0.43 (0.35, 0.54) | |
| p-value | <0.0001 | |
| Overall Survival | ||
| Deaths (%) | 179 (67%) | 206 (79%) |
| Median OS in months (95% CI) | 11.8 (10.5, 13.8) | 6.9 (5.9, 7.6) |
| Hazard ratio(95% CI) | 0.51 (0.41, 0.62) | |
| p-value | <0.0001 | |
Figure 3: Kaplan-Meier Plot of PFS by BICR (All Randomized Patients) in ASCENT
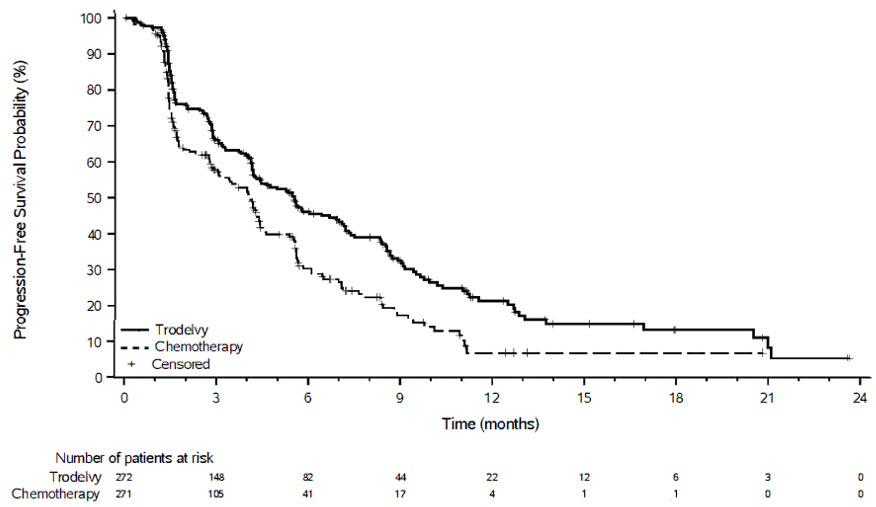
Figure 4: Kaplan-Meier Plot of OS (All Randomized Patients) in ASCENT
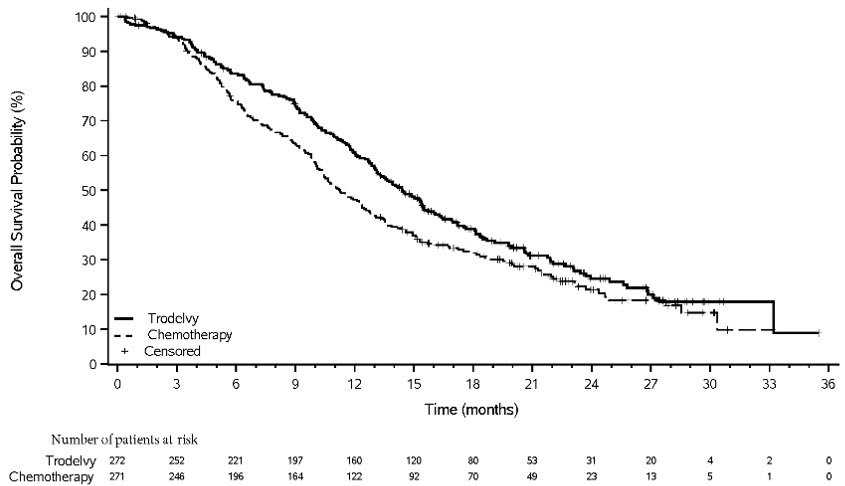
An exploratory analysis of PFS in patients with previously treated, stable brain metastases showed a stratified HR of 0.65 (95% CI: 0.35, 1.22). The median PFS in the TRODELVY arm was 2.8 months (95% CI: 1.5, 3.9) and the median PFS with single agent chemotherapy was 1.6 months (95% CI: 1.3, 2.9). Exploratory OS analysis in the same population showed a stratified HR of 0.87 (95% CI: 0.47, 1.63). The median OS in the TRODELVY arm was 6.8 months (95% CI: 4.7, 14.1) and the median OS with single agent chemotherapy was 7.4 months (95% CI: 4.7, 11.1).
IMMU-132-01
The efficacy of TRODELVY was evaluated in IMMU-132-01 (NCT01631552) a multicenter, single-arm, study that enrolled 108 patients with metastatic TNBC who had received at least two prior anticancer therapies for metastatic disease. Patients with bulky disease, defined as a mass > 7 cm and patients with known Gilbert’s disease were ineligible. Patients with treated brain metastases not receiving high dose steroids (> 20 mg prednisone or equivalent) for at least four weeks were eligible.
Patients received TRODELVY 10 mg/kg via intravenous infusion on Days 1 and 8 of a 21-day cycle.
Assessment of tumor status was performed every 8 weeks, with confirmatory scans obtained 4-6 weeks after an initial partial or complete response. The primary efficacy outcome measure was overall response rate (ORR) per RECIST v1.1. Duration of response (DoR) per RECIST v 1.1 was an additional efficacy outcome.
The median age was 55 years (range: 31 to 80 years); 13% age 65 years or older; 99% female; 76% White, 7% Black, 2.8% Asian, and 0.9% American Indian or Alaska Native; 7% Hispanic/Latino and 93% non-Hispanic/non-Latino; 29% EGOC PS 0 and 71% ECOG PS 1; and 11% had Stage IV disease at the time of initial diagnosis. Seventy-six percent had visceral disease, 42% had hepatic metastases, 56% had lung/pleura metastases, and 2% had brain metastases. The median number of prior systemic therapies received in the metastatic setting was 3 (range: 2 to 10). Prior chemotherapies in the metastatic setting included carboplatin or cisplatin (69%), gemcitabine (55%), paclitaxel or docetaxel (53%), capecitabine (51%), eribulin (45%), doxorubicin (24%), vinorelbine (16%), cyclophosphamide (19%), and ixabepilone (8%). Ninety-eight percent had received prior taxanes and 86% had received prior anthracyclines either in the (neo)adjuvant or metastatic setting.
Table 17 summarizes the efficacy results.
| TRODELVY (N=108) | |
|---|---|
| CI: confidence interval | |
| +: denotes ongoing | |
| Overall Response Rate Investigator assessment | |
| ORR (95% CI) | 33.3% (24.6, 43.1) |
| Complete response | 2.8% |
| Partial response | 30.6% |
| Response duration | |
| Number of responders | 36 |
| Median, Months (95% CI) | 7.7 (4.9, 10.8) |
| Range, Months | 1.9+, 30.4+ |
| % with duration ≥6 months | 55.6% |
| % with duration ≥12 months | 16.7% |
Locally Advanced or Metastatic HR-Positive, HER2-Negative Breast Cancer
TROPiCS-02 Study
The efficacy of TRODELVY was evaluated in TROPiCS-02 (NCT 03901339), a multicenter, open label, randomized study conducted in 543 patients with unresectable locally advanced or metastatic HR-positive, HER2-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer whose disease has progressed after the following in any setting: a CDK 4/6 inhibitor, endocrine therapy, and a taxane. Patients must have received at least two prior chemotherapies in the metastatic setting (one of which could be in the neoadjuvant or adjuvant setting if recurrence occurred within 12 months).
Randomization was stratified by prior chemotherapy regimens for metastatic disease (2 vs. 3-4), visceral metastasis (Yes or No), and endocrine therapy in the metastatic setting for at least 6 months (Yes or No).
Patients were randomized (1:1) to receive one of the following treatment arms:
- TRODELVY 10 mg/kg via an intravenous infusion on Days 1 and 8 of a 21-day cycle (n=272)
- eribulin 1.23 or 1.4 mg/m 2 via intravenous infusion on Days 1 and 8 of a 21-day cycle (n=130), vinorelbine 25 mg/m 2 via intravenous infusion on day 1 of a weekly cycle (n=63), gemcitabine 800 to 1200 mg/m 2 via intravenous infusion on Days 1, 8, and 15 of a 28-day cycle (n=56), or capecitabine 1000 to 1250 mg/m 2 orally twice daily on days 1-14 of a 21-day cycle (n=22).
Assessment of tumor status was performed every 6 weeks for the first 54 weeks followed by every 12 weeks thereafter. Treatment beyond RECIST-defined disease progression was permitted if the patient was clinically stable and considered by the investigator to be deriving clinical benefit. The primary efficacy outcome measure was PFS by BICR per RECIST v1.1. Additional efficacy measures included OS, ORR by BICR, and DOR by BICR.
The median age was 56 years (range: 27–86 years); 26% of patients were 65 years or older; 99% female; 67% White, 3.9% Black, 2.9% Asian, and 26% unknown race; and 45% ECOG PS 0 and 55% ECOG PS 1. Ninety-five percent of patients had visceral metastases. Patients received a median of 7 (range: 3 to 17) prior systemic regimens in any setting and 3 (range: 0 to 8) prior systemic chemotherapy regimens in the metastatic setting. Approximately 42% of patients had 2 prior chemotherapy regimens for treatment of metastatic disease compared to 58% of patients who had 3 to 4 prior chemotherapy regimens. Eighty-six percent of patients received endocrine therapy in the metastatic setting for ³ 6 months.
The trial demonstrated a statistically significant improvement in PFS and OS.
Table 18, Figure 5 and Figure 6 summarize the results of TROPiCS-02.
| All Randomized Patients | ||
|---|---|---|
| TRODELVY n=272 | Single Agent Chemotherapy n=271 | |
| Progression-Free Survival by BICR PFS is defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever comes first. | ||
| Median PFS in months (95% CI) | 5.5 (4.2, 7.0) | 4.0 (3.1, 4.4) |
| Hazard ratio (95% CI) | 0.661 (0.529, 0.826) | |
| p-value Stratified log-rank test adjusted for stratification factors: prior chemotherapy regimens for metastatic disease (2 vs. 3-4), visceral metastasis (Y/N), and endocrine therapy in the metastatic setting for at least 6 months (Yes or No). BICR = Blinded Independent Central Review; CI = Confidence Interval | 0.0003 | |
| Overall Survival Second interim OS analysis (conducted when 390 OS events were observed) | ||
| Median OS in months (95% CI) | 14.4 (13.0, 15.7) | 11.2 (10.1, 12.7) |
| Hazard ratio (95% CI) | 0.789 (0.646, 0.964) | |
| p-value | 0.0200 | |
| Objective Response Rate Objective Response Rate and Duration of response were based on the time of Second interim OS analysis by BICR | ||
| Response Rate, % (95% CI) | 21.0 (16.3, 26.3) | 14.0 (10.1, 18.7) |
| Odds ratio (95% CI) | 1.625 (1.034, 2.555) | |
| p-value | 0.0348 | |
| Duration of Response(DOR) by BICR | ||
| Median DOR in months (95% CI) | 8.1 (6.7, 9.1) | 5.6 (3.8, 7.9) |
Figure 5: Kaplan-Meier Plot of PFS by BICR in TROPiCS-02
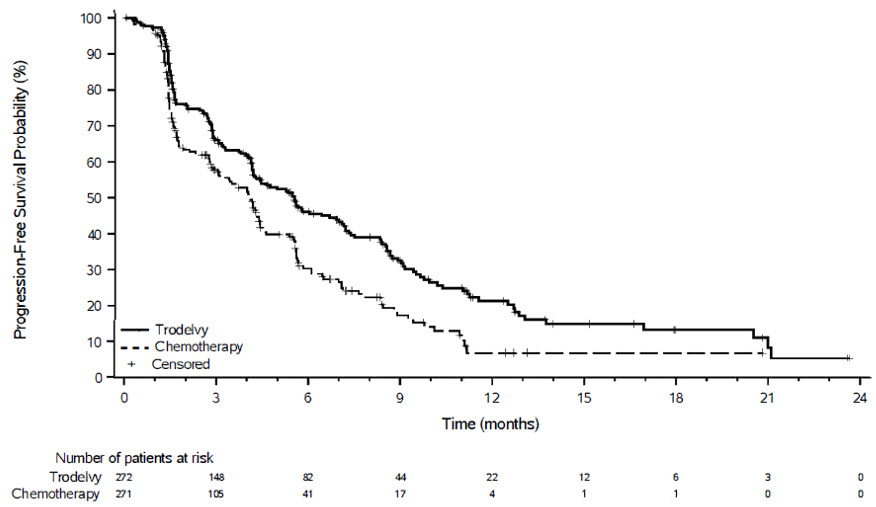
Figure 6: Kaplan-Meier Plot of OS in TROPiCS-02
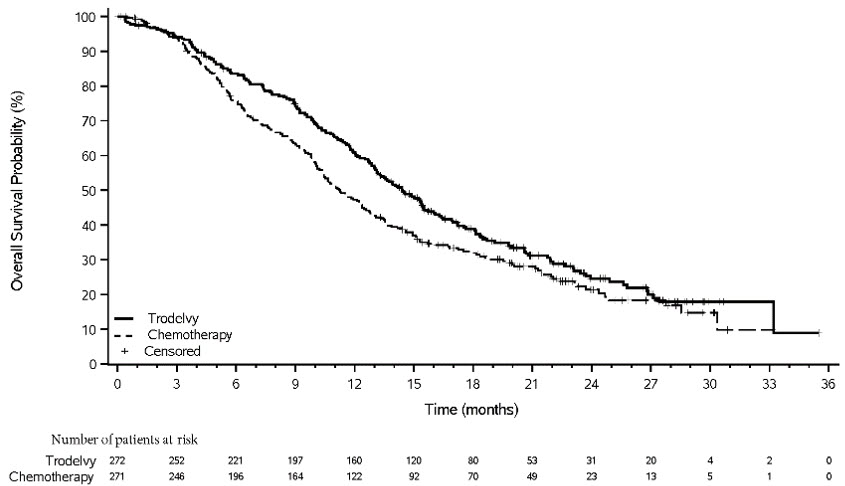
HOW SUPPLIED/STORAGE AND HANDLING
TRODELVY (sacituzumab govitecan-hziy) for injection is a sterile, off-white to yellowish lyophilized powder in a single-dose vial. Each TRODELVY vial is individually boxed in a carton:
- NDC 55135-132-01 contains one 180 mg vial
Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of reconstitution. Do not freeze.
TRODELVY is a hazardous drug. Follow applicable special handling and disposal procedures 1 .
Mechanism of Action
Sacituzumab govitecan-hziy is a Trop-2-directed antibody-drug conjugate. Sacituzumab is a humanized antibody that recognizes Trop-2. The small molecule, SN-38, is a topoisomerase I inhibitor, which is covalently attached to the antibody by a linker. Pharmacology data suggest that sacituzumab govitecan-hziy binds to Trop-2-expressing cancer cells and is internalized with the subsequent release of SN-38 via hydrolysis of the linker. SN-38 interacts with topoisomerase I and prevents re-ligation of topoisomerase I-induced single strand breaks. The resulting DNA damage leads to apoptosis and cell death. Sacituzumab govitecan-hziy decreased tumor growth in mouse xenograft models of triple-negative breast cancer.