Get your patient on Unituxin (Dinutuximab)
Unituxin patient education
Patient toolkit
Dosage & administration

Unituxin prescribing information
WARNING: SERIOUS INFUSION REACTIONS AND NEUROTOXICITY
WARNING: SERIOUS INFUSION REACTIONS AND NEUROTOXICITY
See full prescribing information for complete boxed warning.
- Infusion Reactions: Life-threatening infusion adverse reactions occur with Unituxin. Administer required prehydration and premedication. Immediately interrupt for severe infusion reactions and permanently discontinue for anaphylaxis [seeDosage and Administration (2.2 ,2.3) andWarnings and Precautions (5.1) ] .
- Neurotoxicity: Unituxin causes severe neuropathic pain. Administer intravenous opioid prior to, during, and for 2 hours following completion of the Unituxin infusion. Severe peripheral sensory neuropathy ranged from 2% to 9% in patients with neuroblastoma. Severe peripheral motor neuropathy has also been reported. Discontinue for severe unresponsive pain, severe sensory neuropathy, and moderate to severe peripheral motor neuropathy [seeDosage and Administration (2.2 ,2.3) andWarnings and Precautions (5.2) ] .
Infusion Reactions
- Serious and potentially life-threatening infusion reactions occurred in 26% of patients treated with Unituxin. Administer required prehydration and premedication including antihistamines prior to each Unituxin infusion. Monitor patients closely for signs and symptoms of an infusion reaction during and for at least four hours following completion of each Unituxin infusion. Immediately interrupt Unituxin for severe infusion reactions and permanently discontinue Unituxin for anaphylaxis ( 2.2, 2.3, 5.1 ).
Neurotoxicity
Unituxin causes serious neurologic adverse reactions including severe neuropathic pain and peripheral neuropathy.
Severe neuropathic pain occurs in the majority of patients. Administer intravenous opioid prior to, during, and for 2 hours following completion of the Unituxin infusion.
In clinical studies of patients with high-risk neuroblastoma, Grade 3 peripheral sensory neuropathy occurred in 2% to 9% of patients. In clinical studies of Unituxin and related GD2-binding antibodies, severe motor neuropathy has occurred. Resolution of motor neuropathy did not occur in all cases. Discontinue Unituxin for severe unresponsive pain, severe sensory neuropathy, and moderate to severe peripheral motor neuropathy ( 2.2, 2.3, 5.2 ).
INDICATIONS AND USAGE
Unituxin (dinutuximab) is indicated, in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), and 13-cis-retinoic acid (RA), for the treatment of pediatric patients with high-risk neuroblastoma who achieve at least a partial response to prior first-line multiagent, multimodality therapy [see Clinical Studies (14) ].
DOSAGE AND ADMINISTRATION
- Verify that patients have adequate hematologic, respiratory, hepatic, and renal function prior to initiating each course of Unituxin [see Clinical Studies (14) ].
- Administer required premedication and hydration prior to initiation of each Unituxin infusion [see Dosage and Administration (2.2) ] .
Recommended Dose
- The recommended dose of Unituxin is 17.5 mg/m 2 /day administered as an intravenous infusion over 10 to 20 hours for 4 consecutive days for a maximum of 5 cycles (Table 1 and Table 2) [see Dosage and Administration (2.4) and Clinical Studies (14) ] .
- Initiate at an infusion rate of 0.875 mg/m 2 /hour for 30 minutes. The infusion rate can be gradually increased as tolerated to a maximum rate of 1.75 mg/m 2 /hour. Follow dose modification instructions for adverse reactions [see Dosage and Administration (2.3) ] .
| Cycle Day | 1 through 3 | 4 | 5 | 6 | 7 | 8 through 24 Cycles 1, 3, and 5 are 24 days in duration. |
|---|---|---|---|---|---|---|
| Unituxin | X | X | X | X |
| Cycle Day | 1 through 7 | 8 | 9 | 10 | 11 | 12 through 32 Cycles 2 and 4 are 32 days in duration. |
|---|---|---|---|---|---|---|
| Unituxin | X | X | X | X |
Required Pre-treatment Guidelines
Intravenous Hydration
- Administer 0.9% Sodium Chloride Injection, USP 10 mL/kg as an intravenous infusion over 1 hour just prior to initiating each Unituxin infusion.
Analgesics
- Administer morphine sulfate (50 mcg/kg) intravenously immediately prior to initiation of Unituxin and then continue as a morphine sulfate drip at an infusion rate of 20 to 50 mcg/kg/hour during and for 2 hours following completion of Unituxin.
- Administer additional 25 mcg/kg to 50 mcg/kg intravenous doses of morphine sulfate as needed for pain up to once every 2 hours followed by an increase in the morphine sulfate infusion rate in clinically stable patients.
- Consider using fentanyl or hydromorphone if morphine sulfate is not tolerated.
- If pain is inadequately managed with opioids, consider use of gabapentin or lidocaine in conjunction with intravenous morphine.
Antihistamines and Antipyretics
- Administer an antihistamine such as diphenhydramine (0.5 to 1 mg/kg; maximum dose 50 mg) intravenously over 10 to 15 minutes starting 20 minutes prior to initiation of Unituxin and as tolerated every 4 to 6 hours during the Unituxin infusion.
- Administer acetaminophen (10 to 15 mg/kg; maximum dose 650 mg) 20 minutes prior to each Unituxin infusion and every 4 to 6 hours as needed for fever or pain. Administer ibuprofen (5 to 10 mg/kg) every 6 hours as needed for control of persistent fever or pain.
Dosage Modifications
Manage adverse reactions by infusion interruption, infusion rate reduction, dose reduction, or permanent discontinuation of Unituxin (Table 3 and Table 4) [see Warnings and Precautions (5) , Adverse Reactions (6) , and Clinical Studies (14) ] .
| Grade 3 or 4 anaphylaxis |
| Grade 3 or 4 serum sickness |
| Grade 3 pain unresponsive to maximum supportive measures |
| Grade 4 sensory neuropathy or Grade 3 sensory neuropathy that interferes with daily activities for more than 2 weeks |
| Grade 2 or greater peripheral motor neuropathy |
| Urinary retention that persists following discontinuation of opioids |
| Transverse myelitis |
| Reversible posterior leukoencephalopathy syndrome (RPLS) |
| Subtotal or total vision loss |
| Grade 4 hyponatremia despite appropriate fluid management |
| Infusion-related reactions [see Warnings and Precautions (5.1) ] | |
| Mild to moderate adverse reactions, such as transient rash, fever, rigors, and localized urticaria, that respond promptly to symptomatic treatment | |
| Onset of reaction: | Reduce Unituxin infusion rate to 50% of the previous rate and monitor closely. |
| After resolution: | Gradually increase infusion rate up to a maximum rate of 1.75 mg/m 2 /hour. |
| Prolonged or severe adverse reactions, such as mild bronchospasm without other symptoms, or angioedema that does not affect the airway | |
| Onset of reaction : | Immediately interrupt Unituxin. |
| After resolution: | If signs and symptoms resolve rapidly, resume Unituxin at 50% of the previous rate and observe closely. |
| First recurrence: | Discontinue Unituxin until the following day. If symptoms resolve and continued treatment is warranted, premedicate with hydrocortisone 1 mg/kg (maximum dose 50 mg) intravenously and administer Unituxin at a rate of 0.875 mg/m 2 /hour in an intensive care unit. |
| Second recurrence: | Permanently discontinue Unituxin. |
| Neurological disorders of the eye [see Warnings and Precautions (5.2) ] | |
| Onset of reaction: | Discontinue Unituxin infusion until resolution. |
| After resolution: | Reduce the Unituxin dose by 50%. |
| First recurrence or if accompanied by visual impairment: | Permanently discontinue Unituxin. |
| Capillary leak syndrome [see Warnings and Precautions (5.3) ] | |
| Moderate to severe but not life-threatening capillary leak syndrome | |
| Onset of reaction: | Immediately interrupt Unituxin. |
| After resolution: | Resume Unituxin infusion at 50% of the previous rate. |
| Life-threatening capillary leak syndrome | |
| Onset of reaction: | Discontinue Unituxin for the current cycle. |
| After resolution: | In subsequent cycles, administer Unituxin at 50% of the previous rate. |
| First recurrence: | Permanently discontinue Unituxin. |
| Hypotension Symptomatic hypotension, systolic blood pressure (SBP) less than lower limit of normal for age, or SBP decreased by more than 15% compared to baseline. requiring medical intervention [see Warnings and Precautions (5.4) ] | |
| Onset of reaction: | Interrupt Unituxin infusion. |
| After resolution: | Resume Unituxin infusion at 50% of the previous rate. If blood pressure remains stable for at least 2 hours, increase the infusion rate as tolerated up to a maximum rate of 1.75 mg/m 2 /hour. |
| Severe systemic infection or sepsis [see Warnings and Precautions (5.5) ] | |
| Onset of reaction: | Discontinue Unituxin until resolution of infection, and then proceed with subsequent cycles of therapy. |
Instructions for Preparation and Administration
Preparation
- Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F). Protect from light by storing in the outer carton. DO NOT FREEZE OR SHAKE vials.
- Inspect visually for particulate matter and discoloration prior to administration. Do not administer Unituxin and discard the single-dose vial if the solution is cloudy, has pronounced discoloration, or contains particulate matter.
- Aseptically withdraw the required volume of Unituxin from the single-dose vial and inject into a 100-mL bag of 0.9% Sodium Chloride Injection, USP. Mix by gentle inversion. Do not shake. Discard unused contents of the vial.
- Store the diluted Unituxin solution under refrigeration (2°C to 8°C). Initiate infusion within 4 hours of preparation.
- Discard diluted Unituxin solution 24 hours after preparation.
Administration
- Administer Unituxin as a diluted intravenous infusion only [see Dosage and Administration (2.1) ] . Do not administer Unituxin as an intravenous push or bolus.
DOSAGE FORMS AND STRENGTHS
Injection: 17.5 mg/5 mL (3.5 mg/mL) as a clear and colorless to slightly opalescent solution in a single-dose vial.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Based on its mechanism of action, Unituxin may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1) ] . There are no studies in pregnant women and no reproductive studies in animals to inform the drug-associated risk. Monoclonal antibodies are transported across the placenta in a linear fashion as pregnancy progresses, with the largest amount transferred during the third trimester. Advise pregnant women of the potential risk to a fetus. The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk in the U.S. general population of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies.
Lactation
Risk Summary
There is no information available on the presence of dinutuximab in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. However, human IgG is present in human milk. Because of the potential for serious adverse reactions in a breastfed infant, advise a nursing woman to discontinue breastfeeding during treatment with Unituxin.
Females and Males of Reproductive Potential
Contraception
Females
Unituxin may cause fetal harm [see Use in Specific Populations (8.1) ] . Advise females of reproductive potential to use effective contraception during treatment and for 2 months after the last dose of Unituxin.
Pediatric Use
The safety and effectiveness of Unituxin as part of multi-agent, multimodality therapy have been established in pediatric patients with high-risk neuroblastoma based on results of an open-label, randomized (1:1) trial conducted in 226 patients aged 11 months to 15 years (median age 3.8 years) (Study 1). Prior to enrollment, patients achieved at least a partial response to prior first-line therapy for high-risk neuroblastoma consisting of induction combination chemotherapy, maximum feasible surgical resection, myeloablative consolidation chemotherapy followed by autologous stem cell transplant, and received radiation therapy to residual soft tissue disease. Patients randomized to the Unituxin/13-cis-retinoic acid (RA) arm (Unituxin/RA) received up to 5 cycles of Unituxin in combination with alternating cycles of granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-2 (IL-2) plus RA, followed by 1 cycle of RA alone. Patients randomized to the RA arm received up to 6 cycles of RA monotherapy. Study 1 demonstrated an improvement in event-free survival (EFS) and overall survival (OS) in patients in the Unituxin/RA arm compared to those in the RA arm [see Adverse Reactions (6) , Clinical Pharmacology (12) , and Clinical Studies (14) ] .
Juvenile Animal Toxicity Data
Juvenile monkeys (13 to 18 months of age at study start) received dinutuximab daily via intravenous infusion for 4 consecutive days over five 28-day cycles at doses of 1, 3, or 10 mg/kg. Monkeys also received morphine during infusion for pain management. At the high dose of 10 mg/kg (approximately equal to the 17.5 mg/m 2 clinical dose), mild degeneration of nerve fibers in the brain (medulla oblongata) and moderate degeneration of nerve fibers in the spinal cord (cervical, thoracic, and lumbar) were present. Mild neuronal and nerve fiber degeneration were also present in the dorsal root ganglia (cervical, thoracic, and lumbar). Nerve fiber degeneration in the spinal cord and neuronal degeneration in dorsal root ganglia persisted 6 months after the end of dosing, though at lower severity. At the 10 mg/kg dose level, nerve conduction velocity (NCV) analysis showed motor and sensory NCV decreases of less than 10% compared to vehicle controls, starting on Day 27 and continuing to Day 83. Sensory NCV decreases were still present at the end of the dosing period but were on a trend towards recovery 6 months after the end of dosing.
Geriatric Use
The safety and effectiveness of Unituxin in geriatric patients have not been established.
Renal Impairment
Unituxin has not been studied in patients with renal impairment.
Hepatic Impairment
Unituxin has not been studied in patients with hepatic impairment.
CONTRAINDICATIONS
Unituxin is contraindicated in patients with a history of anaphylaxis to dinutuximab.
WARNINGS AND PRECAUTIONS
- Neurological Disorders of the Eye: Interrupt Unituxin for dilated pupil with sluggish light reflex or other visual disturbances and permanently discontinue Unituxin for recurrent eye disorders or loss of vision. (5.2 )
- Prolonged Urinary Retention and Transverse Myelitis: Permanently discontinue Unituxin and institute supportive care. (5.2 )
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS): Permanently discontinue Unituxin and institute supportive care for signs and symptoms of RPLS. (5.2 )
- Capillary Leak Syndrome and Hypotension: Administer required prehydration and monitor patients closely during treatment. Depending upon severity, manage by interruption, infusion rate reduction, or permanent discontinuation. (5.3 , 5.4 )
- Infection: Interrupt until resolution of systemic infection. (5.5 )
- Bone Marrow Suppression: Monitor peripheral blood counts during Unituxin therapy. (5.6 )
- Electrolyte Abnormalities: Monitor serum electrolytes closely. (5.7 )
- Atypical Hemolytic Uremic Syndrome: Permanently discontinue Unituxin and institute supportive management. (5.8 )
- Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of potential risk to a fetus and to use effective contraception. (5.9 , 8.1 , 8.3 )
Serious Infusion Reactions
Serious infusion reactions requiring urgent intervention, including blood pressure support, bronchodilator therapy, corticosteroids, infusion rate reduction, infusion interruption, or permanent discontinuation of Unituxin, included facial and upper airway edema, dyspnea, bronchospasm, stridor, urticaria, and hypotension. Infusion reactions generally occurred during or within 24 hours of completing the Unituxin infusion. Due to overlapping signs and symptoms, it was not possible to distinguish between infusion reactions and hypersensitivity reactions in some cases.
In Study 1, Severe (Grade 3 or 4) infusion reactions occurred in 35 (26%) patients in the Unituxin/13-cis-retinoic acid (RA) group compared to 1 (1%) patient receiving RA alone. Severe urticaria occurred in 17 (13%) patients in the Unituxin/RA group but did not occur in the RA group. Serious adverse reactions consistent with anaphylaxis and resulting in permanent discontinuation of Unituxin occurred in 2 (1%) patients in the Unituxin/RA group. Additionally, 1 (0.1%) patient had multiple cardiac arrests and died within 24 hours after having received Unituxin in Study 2.
Prior to each Unituxin dose, administer required intravenous hydration and premedication with antihistamines, analgesics, and antipyretics [see Dosage and Administration (2.2) ] . Monitor patients closely for signs and symptoms of infusion reactions during and for at least 4 hours following completion of each Unituxin infusion in a setting where cardiopulmonary resuscitation medication and equipment are available.
For mild to moderate infusion reactions, such as transient rash, fever, rigors, and localized urticaria, that respond promptly to antihistamines or antipyretics, decrease the Unituxin infusion rate and monitor closely. Immediately interrupt or permanently discontinue Unituxin and institute supportive management for severe or prolonged infusion reactions. Permanently discontinue Unituxin and institute supportive management for life-threatening infusion reactions [see Dosage and Administration (2.3) ] .
Neurotoxicity
Pain
In Study 1, 114 (85%) patients treated in the Unituxin/RA group experienced pain despite pre-treatment with analgesics, including morphine sulfate infusion. Severe (Grade 3) pain occurred in 68 (51%) patients in the Unituxin/RA group compared to 5 (5%) patients in the RA group. Pain typically occurred during the Unituxin infusion and was most commonly reported as abdominal pain, generalized pain, extremity pain, back pain, neuralgia, musculoskeletal chest pain, and arthralgia.
Premedicate with analgesics, including intravenous opioids, prior to each dose of Unituxin and continue analgesics until 2 hours following completion of Unituxin [see Dosage and Administration (2.2) ] .
For severe pain, decrease the Unituxin infusion rate to 0.875 mg/m 2 /hour. Discontinue Unituxin if pain is not adequately controlled despite infusion rate reduction and institution of maximum supportive measures [see Dosage and Administration (2.3) ] .
Peripheral Neuropathy
In Study 1, severe (Grade 3) peripheral sensory neuropathy occurred in 2 (1%) patients and severe peripheral motor neuropathy occurred in 2 (1%) patients in the Unituxin/RA group. No patients treated with RA alone experienced severe peripheral neuropathy. The duration and reversibility of peripheral neuropathy occurring in Study 1 was not documented. In Study 3, no patients experienced peripheral motor neuropathy. Among the 9 (9%) patients who experienced peripheral sensory neuropathy of any severity, the median (min, max) duration of peripheral sensory neuropathy was 9 (3, 163) days.
In a study of a related anti-GD2 antibody conducted in 12 adult patients with metastatic melanoma, 2 (13%) patients developed severe motor neuropathy. One patient developed lower extremity weakness and inability to ambulate that persisted for approximately 6 weeks. Another patient developed severe lower extremity weakness resulting in an inability to ambulate without assistance that lasted for approximately 16 weeks and neurogenic bladder that lasted for approximately 3 weeks. Complete resolution of motor neuropathy was not documented in this case.
Permanently discontinue Unituxin in patients with peripheral motor neuropathy of Grade 2 or greater severity, Grade 3 sensory neuropathy that interferes with daily activities for more than 2 weeks, or Grade 4 sensory neuropathy [see Dosage and Administration (2.3) ] .
Neurological Disorders of the Eye
Neurological disorders of the eye experienced by 2 or more patients treated with Unituxin in Studies 1, 2, or 3 included blurred vision, photophobia, mydriasis, fixed or unequal pupils, optic nerve disorder, eyelid ptosis, and papilledema.
In Study 1, 3 (2%) patients in the Unituxin/RA group experienced blurred vision, compared to no patients in the RA group. Diplopia, mydriasis, and unequal pupillary size occurred in 1 patient each in the Unituxin/RA group, compared to no patients in the RA group. The duration of eye disorders occurring in Study 1 was not documented. In Study 3, eye disorders occurred in 16 (15%) patients, and in 3 (3%) patients resolution of the eye disorder was not documented. Among the cases with documented resolution, the median duration of eye disorders was 4 days (range: 0, 221 days).
Interrupt Unituxin in patients experiencing dilated pupil with sluggish light reflex or other visual disturbances that do not cause visual loss. Upon resolution and if continued treatment with Unituxin is warranted, decrease the Unituxin dose by 50%. Permanently discontinue Unituxin in patients with recurrent signs or symptoms of an eye disorder following dose reduction and in patients who experience loss of vision [see Dosage and Administration (2.3) ] .
Prolonged Urinary Retention
Urinary retention that persists for weeks to months following discontinuation of opioids has occurred in patients treated with Unituxin. Permanently discontinue Unituxin in patients with urinary retention that does not resolve following discontinuation of opioids [see Dosage and Administration (2.3) and Postmarketing Experience (6.3) ] .
Transverse Myelitis
Transverse myelitis has occurred in patients treated with Unituxin. Promptly evaluate any patient with signs or symptoms of transverse myelitis, such as weakness, paresthesia, sensory loss, or incontinence. Permanently discontinue Unituxin in patients who develop transverse myelitis [see Dosage and Administration (2.3) and Postmarketing Experience (6.3) ].
Reversible Posterior Leukoencephalopathy Syndrome
Reversible Posterior Leukoencephalopathy Syndrome (RPLS) has occurred in patients treated with Unituxin. Institute appropriate medical treatment and permanently discontinue Unituxin in patients with signs and symptoms of RPLS (eg, severe headache, hypertension, visual changes, lethargy, or seizures) [see Dosage and Administration (2.3) and Postmarketing Experience (6.3) ] .
Capillary Leak Syndrome
In Study 1, severe (Grade 3 to 5) capillary leak syndrome occurred in 31 (23%) patients in the Unituxin/RA group and in no patients treated with RA alone. Additionally, capillary leak syndrome was reported as a serious adverse reaction in 9 (6%) patients in the Unituxin/RA group and in no patients treated with RA alone. Immediately interrupt or discontinue Unituxin and institute supportive management in patients with symptomatic or severe capillary leak syndrome [see Dosage and Administration (2.3) ] .
Hypotension
In Study 1, severe (Grade 3 or 4) hypotension occurred in 22 (16%) patients in the Unituxin/RA group compared to no patients in the RA group.
Prior to each Unituxin infusion, administer required intravenous hydration. Closely monitor blood pressure during Unituxin treatment. Immediately interrupt or discontinue Unituxin and institute supportive management in patients with symptomatic hypotension, systolic blood pressure (SBP) less than lower limit of normal for age, or SBP that is decreased by more than 15% compared to baseline [see Dosage and Administration (2.2 , 2.3) ] .
Infection
In Study 1, severe (Grade 3 or 4) bacteremia requiring intravenous antibiotics or other urgent intervention occurred in 17 (13%) patients in the Unituxin/RA group compared to 5 (5%) patients treated with RA alone. Sepsis occurred in 24 (18%) patients in the Unituxin/RA group and in 10 (9%) patients in the RA group.
Monitor patients closely for signs and symptoms of systemic infection and temporarily discontinue Unituxin in patients who develop systemic infection until resolution of the infection [see Dosage and Administration (2.3) ] .
Bone Marrow Suppression
In Study 1, severe (Grade 3 or 4) thrombocytopenia (39% vs. 25%), anemia (34% vs. 16%), neutropenia (34% vs. 13%), and febrile neutropenia (4% vs. 0 patients) occurred more commonly in patients in the Unituxin/RA group compared to patients treated with RA alone. Monitor peripheral blood counts closely during therapy with Unituxin.
Electrolyte Abnormalities
In Study 1, electrolyte abnormalities occurring in at least 25% of patients who received Unituxin/RA included hyponatremia, hypokalemia, and hypocalcemia. Severe (Grade 3 or 4) hypokalemia and hyponatremia occurred in 37% and 23% of patients in the Unituxin/RA group, respectively, compared to 2% and 4% of patients in the RA group. In a study of a related anti-GD2 antibody conducted in 12 adult patients with metastatic melanoma, 2 (13%) patients developed syndrome of inappropriate antidiuretic hormone secretion resulting in severe hyponatremia. Monitor serum electrolytes daily during therapy with Unituxin.
Atypical Hemolytic Uremic Syndrome
Hemolytic uremic syndrome in the absence of documented infection and resulting in renal insufficiency, electrolyte abnormalities, anemia, and hypertension occurred in 2 patients enrolled in Study 2 following receipt of the first cycle of Unituxin. Atypical hemolytic uremic syndrome recurred following rechallenge with Unituxin in 1 patient. Permanently discontinue Unituxin and institute supportive management for signs of hemolytic uremic syndrome.
Embryo-Fetal Toxicity
Based on its mechanism of action, Unituxin may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment, and for 2 months after the last dose of Unituxin [see Use in Specific Populations (8.1 , 8.3) and Clinical Pharmacology (12.1) ].
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Serious Infusion Reactions [see Boxed Warning and Warnings and Precautions (5.1) ]
- Neurotoxicity, including Pain, Peripheral Neuropathy, Neurological Disorders of the Eye, Prolonged Urinary Retention, Transverse Myelitis, and Reversible Posterior Leukoencephalopathy Syndrome [see Boxed Warning and Warnings and Precautions (5.2) ]
- Capillary Leak Syndrome [see Warnings and Precautions (5.3) ]
- Hypotension [see Warnings and Precautions (5.4) ]
- Infection [see Warnings and Precautions (5.5) ]
- Bone Marrow Suppression [see Warnings and Precautions (5.6) ]
- Electrolyte Abnormalities [see Warnings and Precautions (5.7) ]
- Atypical Hemolytic Uremic Syndrome [see Warnings and Precautions (5.8) ]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.9) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect rates observed in clinical practice.
The data described below reflect exposure to Unituxin at the recommended dose and schedule in 1021 patients with high-risk neuroblastoma enrolled in an open-label, randomized (Study 1), or single-arm clinical trials (Study 2 and Study 3). Prior to enrollment, patients received therapy consisting of induction combination chemotherapy, maximum feasible surgical resection, myeloablative consolidation chemotherapy followed by autologous stem cell transplant, and radiation therapy to residual soft tissue disease. Patients received Unituxin in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), and 13-cis-retinoic acid (RA). Treatment commenced within 95 days post autologous stem cell transplant in Study 1, within 210 days of autologous stem cell transplant in Study 2, and within 110 days of autologous stem cell transplant in Study 3.
Study 1
In a randomized, open-label, multicenter study (Study 1), 134 patients received Unituxin in combination with GM-CSF, IL-2, and RA (Unituxin/RA group), including 109 randomized patients and 25 patients with biopsy-proven residual disease who were non-randomly assigned to receive Unituxin. A total of 106 randomized patients received RA alone (RA group) [see Dosage and Administration (2) and Clinical Studies (14) ] . Patients had a median age at enrollment of 3.8 years (range: 0.94 to 15.3 years), and were predominantly male (60%) and White (82%). In Study 1, adverse reactions of Grade 3 or greater severity were comprehensively collected, but adverse reactions of Grade 1 or 2 severity were collected sporadically and laboratory data were not comprehensively collected.
Approximately 71% of patients in the Unituxin/RA group and 77% of patients in the RA group completed planned treatment. The most common reason for premature discontinuation of study therapy was adverse reactions in the Unituxin/RA group (19%) and progressive disease (17%) in the RA group.
The most common adverse drug reactions (≥25%) in the Unituxin/RA group were pain, pyrexia, thrombocytopenia, lymphopenia, infusion reactions, hypotension, hyponatremia, increased alanine aminotransferase, anemia, vomiting, diarrhea, hypokalemia, capillary leak syndrome, neutropenia, urticaria, hypoalbuminemia, increased aspartate aminotransferase, and hypocalcemia. The most common serious adverse reactions (≥5%) in the Unituxin/RA group were infections, infusion reactions, hypokalemia, hypotension, pain, fever, and capillary leak syndrome.
Table 5 lists the adverse reactions reported in at least 10% of patients in the Unituxin/RA group for which there was a between group difference of at least 5% (all grades) or 2% (Grade 3 or greater severity).
| Adverse Reaction Includes adverse reactions that occurred in at least 10% of patients in the Unituxin/RA group with at least a 5% (All Grades) or 2% (Grades 3 to 5) absolute higher incidence in the Unituxin/RA group compared to the RA group. , Adverse drug reactions were graded using CTCAE version 3.0. | Unituxin/RA (N=134) | RA (N=106) | ||
|---|---|---|---|---|
| All Grades (%) | Grades 3 to 4 (%) | All Grades (%) | Grades 3 to 4 (%) | |
| General Disorders and Administration Site Conditions | ||||
| Pain Includes preferred terms abdominal pain, abdominal pain upper, arthralgia, back pain, bladder pain, bone pain, chest pain, facial pain, gingival pain, infusion related reaction, musculoskeletal chest pain, myalgia, neck pain, neuralgia, oropharyngeal pain, pain, pain in extremity, and proctalgia. | 85 | 51 | 16 | 6 |
| Pyrexia | 72 | 40 | 27 | 6 |
| Edema | 17 | 0 | 0 | 0 |
| Blood and Lymphatic System Disorders Based on investigator reported adverse reactions. | ||||
| Thrombocytopenia | 66 | 39 | 43 | 25 |
| Lymphopenia | 62 | 51 | 36 | 20 |
| Anemia | 51 | 34 | 22 | 16 |
| Neutropenia | 39 | 34 | 16 | 13 |
| Immune System Disorders | ||||
| Infusion reactions | 60 | 25 | 9 | 1 |
| Vascular Disorders | ||||
| Hypotension | 60 | 16 | 3 | 0 |
| Capillary leak syndrome One Grade 5 adverse reaction. | 40 | 23 | 1 | 0 |
| Hemorrhage Includes preferred terms gastrointestinal hemorrhage, hematochezia, rectal hemorrhage, hematemesis, upper gastrointestinal hemorrhage, hematuria, hemorrhage urinary tract, renal hemorrhage, epistaxis, respiratory tract hemorrhage, disseminated intravascular coagulation, catheter site hemorrhage, hemorrhage, and hematoma. | 17 | 6 | 6 | 3 |
| Hypertension | 14 | 2 | 7 | 1 |
| Metabolism and Nutrition Disorders | ||||
| Hyponatremia | 58 | 23 | 12 | 4 |
| Hypokalemia | 43 | 37 | 4 | 2 |
| Hypoalbuminemia | 33 | 7 | 3 | 0 |
| Hypocalcemia | 27 | 7 | 0 | 0 |
| Hypophosphatemia | 20 | 8 | 3 | 0 |
| Hyperglycemia | 18 | 6 | 4 | 1 |
| Hypertriglyceridemia | 16 | 1 | 11 | 1 |
| Decreased appetite | 15 | 10 | 5 | 4 |
| Hypomagnesemia | 12 | 2 | 1 | 0 |
| Investigations | ||||
| Increased alanine aminotransferase | 56 | 23 | 31 | 3 |
| Increased aspartate aminotransferase | 28 | 10 | 7 | 0 |
| Increased serum creatinine | 15 | 2 | 6 | 0 |
| Increased weight | 10 | 0 | 0 | 0 |
| Gastrointestinal Disorders | ||||
| Vomiting | 46 | 6 | 19 | 3 |
| Diarrhea | 43 | 13 | 15 | 1 |
| Nausea | 10 | 2 | 3 | 1 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Urticaria | 37 | 13 | 3 | 0 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Hypoxia | 24 | 12 | 2 | 1 |
| Cardiac Disorders | ||||
| Tachycardia Includes preferred terms tachycardia and sinus tachycardia. | 19 | 2 | 1 | 0 |
| Infections and Infestations | ||||
| Sepsis | 18 | 16 | 9 | 9 |
| Device related infection | 16 | 16 | 11 | 11 |
| Renal and Urinary Disorders | ||||
| Proteinuria | 16 | 0 | 3 | 1 |
| Nervous System Disorders | ||||
| Peripheral neuropathy | 13 | 3 | 6 | 0 |
Table 6 compares the per-patient incidence of selected adverse reactions occurring during cycles containing Unituxin in combination with GM-CSF (Cycles 1, 3, and 5) with cycles containing Unituxin in combination with IL-2 (Cycles 2 and 4).
| Preferred Term Includes preferred terms with a per-patient incidence of at least 20% in the Unituxin and RA group for either IL-2 or GM-CSF containing cycles. , Adverse drug reactions were graded using CTCAE version 3.0. | All Grades | Severe | ||
|---|---|---|---|---|
| GM-CSF N=134 (%) | IL-2 Seven patients who received GM-CSF in Cycle 1 discontinued prior to starting Cycle 2. N=127 (%) | GM-CSF N=134 (%) | IL-2 N=127 (%) | |
| GM-CSF, granulocyte-macrophage colony-stimulating factor; IL-2, interleukin-2; RA, 13-cis-retinoic acid | ||||
| General Disorders and administration site conditions | ||||
| Pyrexia | 55 | 65 | 10 | 37 |
| Pain Includes preferred terms abdominal pain, abdominal pain upper, arthralgia, back pain, bladder pain, bone pain, chest pain, facial pain, gingival pain, infusion related reaction, musculoskeletal chest pain, myalgia, neck pain, neuralgia, oropharyngeal pain, pain, pain in extremity, and proctalgia. | 77 | 61 | 43 | 35 |
| Blood and Lymphatic System Disorders Based on investigator-reported adverse reactions. | ||||
| Thrombocytopenia | 62 | 61 | 31 | 33 |
| Lymphopenia | 54 | 61 | 33 | 50 |
| Anemia | 42 | 42 | 21 | 24 |
| Neutropenia | 25 | 32 | 19 | 28 |
| Immune System Disorders | ||||
| Infusion reactions | 47 | 54 | 10 | 20 |
| Vascular Disorders | ||||
| Hypotension | 43 | 54 | 5 | 16 |
| Capillary leak syndrome | 22 | 36 | 11 | 20 |
| Metabolism and Nutrition Disorders | ||||
| Hyponatremia | 36 | 55 | 5 | 21 |
| Hypokalemia | 26 | 39 | 13 | 33 |
| Hypoalbuminemia | 29 | 29 | 3 | 5 |
| Hypocalcemia | 20 | 21 | 2 | 6 |
| Investigations | ||||
| Increased alanine aminotransferase | 43 | 48 | 15 | 13 |
| Aspartate aminotransferase increased | 16 | 21 | 4 | 7 |
| Gastrointestinal Disorders | ||||
| Diarrhea | 31 | 37 | 6 | 13 |
| Vomiting | 33 | 35 | 3 | 2 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Urticaria | 25 | 29 | 7 | 7 |
Study 2 and Study 3
Study 2 was a single-arm, multicenter, expanded access trial that enrolled patients with high-risk neuroblastoma (N=783). The reported adverse event profile of Unituxin in Study 2 was similar to that observed in Study 1.
Study 3 was a multicenter, single-arm safety study of Unituxin in combination with GM-CSF, IL-2, and RA. In Study 3, adverse events of all CTCAE grades and laboratory data were systematically and comprehensively collected. Of 104 patients enrolled and treated in Study 3, 77% of patients completed study therapy. In general, the adverse reaction profile of Unituxin observed in Study 3 was similar to that observed in Study 1 and Study 2. The following adverse reactions not previously reported in Study 1 were reported in at least 10% of patients in Study 3: nasal congestion (20%) and wheezing (15%). Table 7 provides the per-patient incidence of laboratory abnormalities in Study 3.
| Laboratory Test Laboratory abnormalities with a per-patient incidence of at least 20% (all grades) and at least a 5% per-patient incidence of severe (Grade 3 or 4) laboratory abnormalities. | Grade Based on CTCAE version 4.0. | |
|---|---|---|
| All Grades % | Grades 3 to 4 % | |
| ND, not determined | ||
| Hematology | ||
| Anemia | 100 | 46 |
| Neutropenia | 99 | 63 |
| Thrombocytopenia | 98 | 49 |
| Chemistry | ||
| Hypoalbuminemia | 100 | 8 |
| Hypocalcemia | 97 | 7 |
| Hyponatremia | 93 | 36 |
| Hyperglycemia | 87 | 6 |
| Aspartate Aminotransferase Increased | 84 | 8 |
| Alanine Aminotransferase Increased | 83 | 13 |
| Hypokalemia | 82 | 41 |
| Hypophosphatemia | 78 | 6 |
| Urinalysis Urinalysis results were reported as positive or negative without assessment of grade. | ||
| Urine protein | 66 | ND |
| Red blood cell casts | 38 | ND |
Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay.
Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Among the 418 patients who were treated with Unituxin in combination with GM-CSF, IL-2, and RA in Study 2, Study 3 and Study DIV-NB-201, 86 patients (20.6%) tested positive for anti-dinutuximab antibodies (ADA). Of the 86 ADA positive patients, 45 (52.3%) tested positive for neutralizing antibodies.
Among the 27 patients who were treated with Unituxin in combination with lenalidomide and isotretinoin in Study NANT 2011-04, 13 patients (48.1%) tested positive for ADA. Of the 13 ADA positive patients, 2 (15.4%) also tested positive for neutralizing antibodies.
The clearance of dinutuximab was 60% higher for the ADA positive patients than the ADA negative patients. The presence of ADA did not have a clinically significant effect on the incidence or severity of adverse reactions. There were insufficient number of patients with ADA to determine whether ADA alters the efficacy of Unituxin.
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of Unituxin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Neurotoxicity: prolonged urinary retention, transverse myelitis, and reversible posterior leukoencephalopathy syndrome (RPLS) [see Warnings and Precautions (5.2) ] .
DRUG INTERACTIONS
No drug-drug interaction studies have been conducted with dinutuximab.
DESCRIPTION
Dinutuximab is a glycolipid disialoganglioside (GD2)-binding chimeric monoclonal antibody composed of murine variable heavy and light chain regions and the human constant region for the heavy chain IgG 1 and light chain kappa. Dinutuximab is produced in the murine myeloma cell line, SP2/0 and has an approximate molecular weight of 150 kDa.
Unituxin (dinutuximab) injection is a sterile, preservative-free, clear and colorless to slightly opalescent solution for intravenous infusion. Unituxin is supplied in single-dose vials of 17.5 mg/5 mL. Each mL of solution contains 3.5 mg of dinutuximab, and histidine (3.10 mg), polysorbate 20 (0.55 mg), sodium chloride (8.77 mg), and Water for Injection, USP; hydrochloric acid is added to adjust pH to 6.8.
CLINICAL PHARMACOLOGY
Mechanism of Action
Dinutuximab binds to the glycolipid GD2. This glycolipid is expressed on neuroblastoma cells and on normal cells of neuroectodermal origin, including the central nervous system and peripheral nerves. Dinutuximab binds to cell surface GD2 and induces cell lysis of GD2-expressing cells through antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).
Pharmacokinetics
The pharmacokinetics of dinutuximab was evaluated by a population pharmacokinetic analysis in a clinical study of Unituxin in combination with GM-CSF, IL-2, and RA. In this study, 27 children with high-risk neuroblastoma (age: 3.9±1.9 years) received up to 5 cycles of Unituxin at 17.5 mg/m 2 /day as an intravenous infusion over 10 to 20 hours for 4 consecutive days every 28 days. The observed maximum plasma dinutuximab concentration (C max ) was 11.5 mcg/mL (20%, coefficient of variation [CV]). The mean volume of distribution at steady state (Vd ss ) was 5.4 L (28%). The clearance was 0.21 L/day (62%) and increased with body size. The terminal half-life was 10 days (56%).
No formal pharmacokinetic studies were conducted in patients with renal or hepatic impairment.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been conducted to evaluate the carcinogenic or mutagenic potential of dinutuximab.
Dedicated studies examining the effects of dinutuximab on fertility in animals have not been conducted. No clear effects on reproductive organs were observed in general toxicology studies conducted in rats.
Animal Toxicology and/or Pharmacology
Nonclinical studies suggest that dinutuximab-induced neuropathic pain and neurotoxicity are mediated by binding of the antibody to the GD2 antigen located on the surface of nerve fibers and myelin and subsequent induction of CDC and ADCC activity.
CLINICAL STUDIES
The safety and effectiveness of Unituxin was evaluated in a randomized, open-label, multicenter trial conducted in pediatric patients with high-risk neuroblastoma (Study 1). All patients had received prior therapy consisting of induction combination chemotherapy, maximum feasible surgical resection, myeloablative consolidation chemotherapy followed by autologous stem cell transplant, and radiation therapy to residual soft tissue disease. Patients were randomized between Day 50 and Day 77 post-autologous stem cell transplantation.
Patients were required to have achieved at least a partial response prior to autologous stem cell transplantation, have no evidence of disease progression following completion of front-line multi-modality therapy, have adequate pulmonary function (no dyspnea at rest and peripheral arterial oxygen saturation of at least 94% on room air), adequate hepatic function (total bilirubin <1.5× the upper limit of normal and ALT <5× the upper limit of normal), adequate cardiac function (shortening fraction of >30% by echocardiogram, or if shortening fraction abnormal, ejection fraction of 55% by gated radionuclide study), and adequate renal function (glomerular filtration rate at least 70 mL/min/1.73 m 2 ). Patients with systemic infections or a requirement for concomitant systemic corticosteroids or immunosuppressant usage were not eligible for enrollment.
Patients randomized to the Unituxin/RA arm received up to 5 cycles of Unituxin (clinical trials material) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) (Table 8) or interleukin-2 (IL-2) (Table 9) plus 13-cis-retinoic acid (RA), followed by 1 cycle of RA alone. Patients randomized to the RA arm received 6 cycles of RA. Unituxin was administered at a dose of 17.5 mg/m 2 /day (equivalent to 25 mg/m 2 /day of clinical trials material) on 4 consecutive days. Patients in both treatment arms received 6 cycles of RA at a dose of 160 mg/m 2 /day orally (for patients weighing more than 12 kg) or 5.33 mg/kg/day (for patients weighing less than or equal to 12 kg) in 2 divided doses for 14 consecutive days.
| Cycle Day | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15-24 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GM-CSF GM-CSF: 250 µg/m 2 /day, administered by either subcutaneous injection (recommended) or IV infusion administered over 2 hours. | X | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Unituxin Unituxin: 17.5 mg/m 2 /day, administered by diluted IV infusion over 10 to 20 hours. | X | X | X | X | |||||||||||
| RA RA: for >12 kg body weight, 80 mg/m 2 orally twice daily for a total dose of 160 mg/m 2 /day; for ≤12 kg body weight, 2.67 mg/kg orally twice daily for a total daily dose of 5.33 mg/kg/day (round dose up to nearest 10 mg). | X | X | X | X | X |
| Cycle Day | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12-14 | 15-28 | 29-32 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL-2 IL-2: 3 MIU/m 2 /day administered by continuous IV infusion over 96 hours on Days 1 to 4 and 4.5 MIU/m 2 /day on Days 8 to 11. | X | X | X | X | X | X | X | X | ||||||
| Unituxin Unituxin: 17.5 mg/m 2 /day, administered by diluted IV infusion over 10 to 20 hours. | X | X | X | X | ||||||||||
| RA RA: for >12 kg body weight, 80 mg/m 2 orally twice daily for a total dose of 160 mg/m 2 /day; for ≤12 kg body weight, 2.67 mg/kg orally twice daily for a total daily dose of 5.33 mg/kg/day (round dose up to nearest 10 mg). | X |
A total of 226 patients were randomized, 113 patients to each arm. In general, demographic and baseline tumor characteristics were similar across study arms. Across the study population, 60% were male, the median age was 3.8 years and 3% of patients were less than 1.5 years, 82% were White and 7% were Black. The majority (80%) of patients had International Neuroblastoma Staging System Stage 4 disease. Thirty-five percent of patients had a complete response, 43% had a very good partial response, and 23% had a partial response to therapy received prior to autologous stem cell transplant. Forty-six percent of patients had neuroblastoma that was not MYCN-amplified, 36% had tumors with known MYCN-amplification, and MYCN status was unknown or missing in 19% of patients. Forty-three percent of patients had hyperdiploid tumors, 36% had diploid tumors, and DNA ploidy status was unknown or missing in 21% of patients.
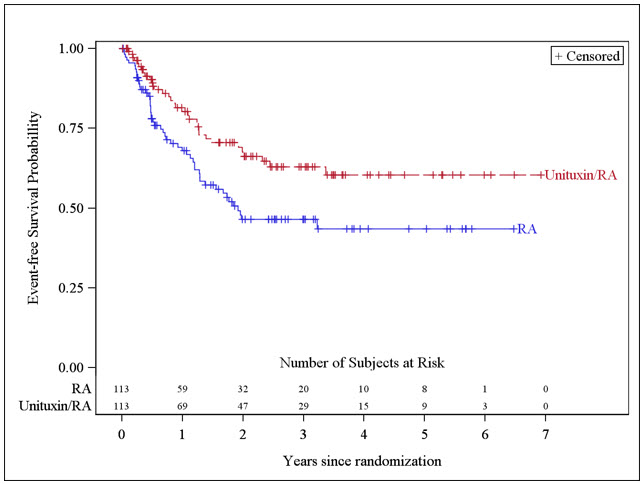
The major efficacy outcome measure was investigator-assessed event-free survival (EFS), defined as the time from randomization to the first occurrence of relapse, progressive disease, secondary malignancy, or death. Overall survival (OS) was also evaluated. After observing a numerical improvement in EFS based on the seventh interim analysis, the Data Monitoring Committee recommended termination of accrual. Efficacy results are shown in Table 10.
| Efficacy Parameter | Unituxin/RA arm n=113 | RA arm n=113 | |
|---|---|---|---|
| NR, not reached | |||
| EFS | No. of Events (%) | 33 (29%) | 50 (44%) |
| Median (95% CI) (years) | NR (3.4, NR) | 1.9 (1.3, NR) | |
| Hazard Ratio (95% CI) | 0.57 (0.37, 0.89) | ||
| p-value (log-rank test) Compared to the allocated alpha of 0.01 pre-specified for the seventh interim analysis of EFS | 0.01 | ||
| OS Based on an additional 3 years of follow up after the seventh interim analysis of EFS | No. of Events (%) | 31 (27%) | 48 (42%) |
| Median (95% CI) (years) | NR (7.5, NR) | NR (3.9, NR) | |
| Hazard Ratio (95% CI) | 0.58 (0.37, 0.91) | ||
The Kaplan-Meier curve of EFS is shown in Figure 1.
Figure 1: Kaplan-Meier Curve of Event-Free Survival
HOW SUPPLIED/STORAGE AND HANDLING
Unituxin (dinutuximab) injection as a clear and colorless to slightly opalescent solution is supplied in a carton containing one 17.5 mg/5 mL (3.5 mg/mL) single-dose vial.
NDC 66302-014-01
Store Unituxin vials under refrigeration at 2°C to 8°C (36°F to 46°F) in the outer carton to protect from light until time of use. Do not freeze or shake the vial.
Mechanism of Action
Dinutuximab binds to the glycolipid GD2. This glycolipid is expressed on neuroblastoma cells and on normal cells of neuroectodermal origin, including the central nervous system and peripheral nerves. Dinutuximab binds to cell surface GD2 and induces cell lysis of GD2-expressing cells through antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).