Get your patient on Vanflyta (Quizartinib)
Vanflyta patient education
Patient toolkit
Dosage & administration

Vanflyta prescribing information
WARNING: QT PROLONGATION, TORSADES DE POINTES, and CARDIAC ARREST
- VANFLYTA prolongs the QT interval in a dose- and concentration-related manner [see Clinical Pharmacology (12.2) ] . Prior to VANFLYTA administration and periodically, monitor for hypokalemia or hypomagnesemia, and correct deficiencies. Perform ECGs to monitor the QTc at baseline, weekly during induction and consolidation therapy, weekly for at least the first month of maintenance, and periodically thereafter [see Dosage and Administration (2.3) and Warnings and Precautions (5.1) ].
- Torsades de pointes and cardiac arrest have occurred in patients receiving VANFLYTA. Do not administer VANFLYTA to patients with severe hypokalemia, severe hypomagnesemia, or long QT syndrome [see Contraindications (4) and Warnings and Precautions (5.1) ] .
- Do not initiate treatment with VANFLYTA or escalate the VANFLYTA dose if the QT interval corrected by Fridericia's formula (QTcF) is greater than 450 ms [see Dosage and Administration (2.3) and Warnings and Precautions (5.1) ] .
- Monitor ECGs more frequently if concomitant use of drugs known to prolong the QT interval is required [see Dosage and Administration (2.3) and Warnings and Precautions (5.1) ] .
- Reduce the VANFLYTA dose when used concomitantly with strong CYP3A inhibitors, as they may increase quizartinib exposure [see Dosage and Administration (2.4) and Warnings and Precautions (5.1) ] .
- Because of the risk of QT prolongation, VANFLYTA is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the VANFLYTA REMS [see Warnings and Precautions (5.2) ] .
INDICATIONS AND USAGE
VANFLYTA is indicated in combination with standard cytarabine and anthracycline induction and cytarabine consolidation, and as maintenance monotherapy following consolidation chemotherapy, for the treatment of adult patients with newly diagnosed acute myeloid leukemia (AML) that is FLT3 internal tandem duplication (ITD)-positive as detected by an FDA-approved test [see Dosage and Administration (2.1) and Clinical Studies (14) ] .
Limitations of Use
VANFLYTA is not indicated as maintenance monotherapy following allogeneic hematopoietic stem cell transplantation (HSCT); improvement in overall survival with VANFLYTA in this setting has not been demonstrated [see Clinical Studies (14) ] .
DOSAGE AND ADMINISTRATION
Patient Selection
Select patients for the treatment of AML with VANFLYTA based on the presence of FLT3-ITD mutation positivity [see Clinical Studies (14) ] . Information on FDA-approved tests for the detection of FLT3-ITD mutation in AML is available at: http://www.fda.gov/CompanionDiagnostics.
Recommended Dosage
- A treatment course consists of up to 2 cycles of VANFLYTA in combination with induction cytarabine and anthracycline, up to 4 cycles of VANFLYTA in combination with high-dose cytarabine consolidation, and up to 36 cycles of VANFLYTA as maintenance therapy [see Clinical Studies (14) ] or until disease progression or unacceptable toxicity. VANFLYTA maintenance therapy should be initiated following consolidation chemotherapy upon blood count recovery of absolute neutrophil count >500/mm 3 and platelet count >50,000/mm 3 .
- See Table 1 for the recommended dosage of VANFLYTA by phase of therapy .
| VANFLYTA Initiation | Induction Patients can receive up to 2 cycles of induction. | Consolidation Patients can receive up to 4 cycles of consolidation. | Maintenance |
|---|---|---|---|
| Starting on Day 8 (for 7 + 3 regimen) For 5 + 2 regimen as the second induction cycle, VANFLYTA will be given on Days 6 to 19. | Starting on Day 6 | Starting on Day 1 | |
| Dose | 35.4 mg orally once daily | 35.4 mg orally once daily |
|
| Duration (28-day cycles) | Two weeks in each cycle (Days 8 to 21) | Two weeks in each cycle (Days 6 to 19) |
|
For patients who proceed to hematopoietic stem cell transplantation (HSCT), VANFLYTA should be stopped 7 days before the start of a conditioning regimen.
Administer VANFLYTA orally with or without food at approximately the same time each day. Swallow tablets whole. Do not cut, crush, or chew the tablets. If a dose of VANFLYTA is vomited, do not administer a replacement dose; wait until the next scheduled dose is due. If a dose of VANFLYTA is missed or not taken at the usual time, administer the dose as soon as possible on the same day and return to the usual schedule the following day. The patient should not take two doses on the same day.
Monitoring and Dosage Modifications for Adverse Reactions
Initiate VANFLYTA only if QTcF is less than or equal to 450 ms [see Warnings and Precautions (5.1) ].
During induction and consolidation, perform ECGs prior to initiation and then once weekly during VANFLYTA treatment or more frequently as clinically indicated [see Warnings and Precautions (5.1) ].
During maintenance, perform ECGs prior to initiation, once weekly for at least the first month following dose initiation and escalation, and thereafter as clinically indicated. Escalate the dose only if QTcF is less than or equal to 450 ms [see Dosage and Administration (2.2) and Warnings and Precautions (5.1) ].
Correct electrolyte abnormalities (hypokalemia and hypomagnesemia), and if possible, avoid concomitant administration of drugs that prolong the QT interval [see Warnings and Precautions (5.1) ].
For recommended dosage modifications due to adverse reactions, see Table 2 . For dosage adjustments due to adverse reactions, see Table 3 .
| Adverse Reaction | Recommended Action |
|---|---|
| Grades are in accordance with National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 (NCI CTCAE v4.03). | |
| QTcF between 450 ms and 480 ms (Grade 1) |
|
| QTcF between 481 ms and 500 ms (Grade 2) |
|
| QTcF greater than 500 ms (Grade 3) |
|
| Recurrent QTcF greater than 500 ms (Grade 3) |
|
| Torsades de pointes, polymorphic ventricular tachycardia, signs/symptoms of life-threatening arrhythmia (Grade 4) |
|
| Grade 3 or 4 non-hematologic adverse reactions |
|
| Grade 3 or 4 hypokalemia (<3 mmol/L) or hypomagnesemia (<0.4 mmol/L or <0.9 mg/dL) |
|
| Grade 4 neutropenia or thrombocytopenia after achieving remission Recommend bone marrow evaluation. |
|
| Current Dosage | Modified Dosage |
|---|---|
| 53 mg once daily | 35.4 mg once daily |
| 35.4 mg once daily | 26.5 mg once daily |
| 26.5 mg once daily | Interrupt |
| 17.7 mg once daily | Interrupt |
Dosage Modifications for Strong CYP3A Inhibitors
Reduce the dosage of VANFLYTA when used concomitantly with strong CYP3A inhibitors as shown in Table 4. If the current dosage is 17.7 mg once daily, interrupt VANFLYTA treatment for the duration of strong CYP3A inhibitor use. After discontinuation of a strong CYP3A inhibitor for 5 half-lives, resume the VANFLYTA dose that was taken before initiating the strong inhibitor [see Drug Interactions (7) ] .
| Current Dosage | Modified Dosage |
|---|---|
| 53 mg once daily | 26.5 mg once daily |
| 35.4 mg once daily | 17.7 mg once daily |
| 26.5 mg once daily | 17.7 mg once daily |
DOSAGE FORMS AND STRENGTHS
Tablets:
- 17.7 mg quizartinib, white, round, film-coated, debossed with "DSC511"
- 26.5 mg quizartinib, yellow, round, film-coated, debossed with "DSC512"
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2 )
Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, VANFLYTA can cause embryo-fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1) ] .
There are no available data on VANFLYTA use in pregnant women to evaluate for a drug-associated risk. In animal reproduction studies, oral administration of quizartinib to pregnant rats during organogenesis resulted in adverse developmental outcomes including structural abnormalities and alterations to growth at maternal exposures approximately 3 times those in patients at the maximum recommended human dose (MRHD) of 53 mg/day ( see Data ). Advise pregnant women of the potential risk to a fetus.
The background risk in the U.S. general population of major birth defects is 2-4%, and of miscarriage is 15-20% of clinically recognized pregnancies.
Data
Animal Data
In an embryo-fetal development study in rats, pregnant animals received oral doses of quizartinib of 0, 0.6, 2, or 6 mg/kg/day during the period of organogenesis. Administration of quizartinib at the dose of 6 mg/kg/day was associated with adverse developmental outcomes including structural abnormalities (anasarca and edema) and alterations to growth (lower fetal weights and effects on skeletal ossification). At this dose, the maternal systemic exposures (AUC) were approximately 3 times the human exposure at the MRHD of 53 mg/day.
Lactation
Risk Summary
There are no data on the presence of quizartinib or its metabolites in human milk, or the effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with VANFLYTA and for one month after the last dose.
Females and Males of Reproductive Potential
VANFLYTA can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1) ] .
Pregnancy Testing
Verify pregnancy status in females of reproductive potential within seven days before starting treatment with VANFLYTA.
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during treatment with VANFLYTA and for 7 months after the last dose.
Males
Based on genotoxicity findings, advise male patients with female partners of reproductive potential to use effective contraception during treatment with VANFLYTA and for 4 months after the last dose [see Nonclinical Toxicology (13.1) ] .
Infertility
Females
Based on findings from animal studies, VANFLYTA may impair female fertility. These effects on fertility were reversible [see Nonclinical Toxicology (13.1) ] .
Males
Based on findings from animal studies, VANFLYTA may impair male fertility. These effects on fertility were reversible [see Nonclinical Toxicology (13.1) ].
Pediatric Use
Safety and effectiveness of VANFLYTA have not been established in pediatric patients.
Geriatric Use
There were 533 patients with newly diagnosed AML in the clinical study; of the total number of VANFLYTA-treated patients, 69 (26%) were 65 years of age and older, while 1 (0.4%) was 75 years of age [see Clinical Studies (14) ] . No overall differences in safety or efficacy were observed between patients 65 years of age and older and younger adult patients.
Renal Impairment
No dosage adjustment is recommended in patients with mild to moderate renal impairment (i.e., estimated creatinine clearance [CLcr] by Cockcroft-Gault equation: CLcr 30 to 89 mL/min). VANFLYTA has not been studied in patients with severe renal impairment (CLcr <30 mL/min) [see Clinical Pharmacology (12.3) ] .
Hepatic Impairment
No dosage adjustment is recommended in patients with mild hepatic impairment (Child-Pugh Class A or total bilirubin ≤ upper limit of normal [ULN] and aspartate aminotransferase [AST] >ULN, or total bilirubin >1 to 1.5 times ULN and any value for AST) or moderate hepatic impairment (Child-Pugh Class B or total bilirubin >1.5 to 3 times ULN and any value for AST). VANFLYTA has not been studied in patients with severe (Child-Pugh Class C or total bilirubin >3 times ULN and any value for AST) hepatic impairment [see Clinical Pharmacology (12.3) ] .
CONTRAINDICATIONS
VANFLYTA is contraindicated in patients with severe hypokalemia, severe hypomagnesemia, long QT syndrome, or in patients with a history of ventricular arrhythmias or torsades de pointes [see Warnings and Precautions (5.1) ].
WARNINGS AND PRECAUTIONS
- QT Prolongation, Torsades de Pointes, and Cardiac Arrest: Monitor electrocardiograms and levels of serum electrolytes. Reduce, interrupt, or permanently discontinue VANFLYTA as appropriate. (2.3 , 5.1 )
- Embryo-Fetal Toxicity: VANFLYTA can cause fetal harm. Advise females of reproductive potential and males with female partners of reproductive potential of potential risk to a fetus and to use effective contraception. (5.3 , 8.1 , 8.3 )
QT Prolongation, Torsades de Pointes, and Cardiac Arrest
VANFLYTA prolongs the QT interval in a dose- and concentration-dependent manner. The mechanism of QTc interval prolongation is via inhibition of the slow delayed rectifier potassium current, I Ks , as compared to all other medications that prolong the QTc interval, which is via the rapid delayed rectifier potassium current, I Kr . Therefore, the level of QTc prolongation with VANFLYTA that predicts the risk of cardiac arrhythmias is unclear. Inhibition of I Ks and I Kr may leave patients with limited reserve leading to a higher risk of QT prolongation and serious cardiac arrhythmias, including fatal outcomes [see Clinical Pharmacology (12.2) ] . Torsades de pointes, ventricular fibrillation, cardiac arrest, and sudden death have occurred in patients treated with VANFLYTA.
Of the 1,081 patients with AML treated with VANFLYTA in clinical trials, torsades de pointes occurred in approximately 0.2% of patients, cardiac arrest occurred in 0.6%, including 0.4% with a fatal outcome, and 0.1% of patients experienced ventricular fibrillation [see Adverse Reactions (6.1) ] . These severe cardiac arrhythmias occurred predominantly during the induction phase.
Of the 265 patients with newly diagnosed FLT3-ITD-positive AML treated with VANFLYTA in combination with chemotherapy in the clinical trial, 2.3% were found to have a QTcF greater than 500 ms and 10% of patients had an increase from baseline QTcF greater than 60 ms. The clinical trial excluded patients with a QTcF ≥450 ms or other factors that increased the risk of QT prolongation or arrhythmic events (e.g., NYHA Class III or IV congestive heart failure, hypokalemia, family history of long QT interval syndrome). Therefore, avoid use in patients who are at significant risk of developing torsades de pointes, including uncontrolled or significant cardiac disease, recent myocardial infarction, heart failure, unstable angina, bradyarrhythmias, tachyarrhythmias, uncontrolled hypertension, high-degree atrioventricular block, severe aortic stenosis, or uncontrolled hypothyroidism.
Do not initiate treatment with VANFLYTA if the QTcF interval is greater than 450 ms. Do not use VANFLYTA in patients with severe hypokalemia, severe hypomagnesemia, long QT syndrome, or in patients with a history of ventricular arrhythmias or torsades de pointes [see Contraindications (4) ] .
Perform an ECG and correct electrolyte abnormalities prior to initiation of treatment with VANFLYTA. During induction and consolidation, perform an ECG prior to initiation and then once weekly during VANFLYTA treatment or more frequently as clinically indicated. During maintenance, perform ECGs prior to initiation, once weekly for at least the first month following dose initiation and escalation, and as clinically indicated thereafter. Do not escalate the dose if QTcF is greater than 450 ms [see Dosage and Administration (2.3) ] .
Perform ECG monitoring of the QT interval more frequently in patients who are at significant risk of developing QT interval prolongation and torsades de pointes, or following dose escalation.
Monitor and correct hypokalemia and hypomagnesemia prior to and during treatment with VANFLYTA. Maintain electrolytes in the normal range. Monitor electrolytes and ECGs more frequently in patients who experience diarrhea or vomiting.
Monitor patients more frequently with ECGs if coadministration of VANFLYTA with drugs known to prolong the QT interval is required [see Drug Interactions (7) ] .
Reduce the VANFLYTA dose when used concomitantly with strong CYP3A inhibitors, as they may increase quizartinib exposure [see Dosage and Administration (2.4) ] .
Reduce VANFLYTA if QTc increases to greater than 480 ms and less than 500 ms. Interrupt and reduce VANFLYTA if QTc increases to greater than 500 ms. Permanently discontinue VANFLYTA in patients who develop recurrent QTc greater than 500 ms or QTc interval prolongation with signs or symptoms of life-threatening arrhythmia [see Dosage and Administration (2.3) ] .
VANFLYTA is available only through a restricted program under a REMS [see Warnings and Precautions (5.2) ] .
VANFLYTA REMS
VANFLYTA is available only through a restricted distribution program under a REMS called the VANFLYTA REMS because of the serious risk of QT prolongation, torsades de pointes, and cardiac arrest [see Warnings and Precautions (5.1) ] .
Notable requirements of the VANFLYTA REMS include the following:
- Prescribers must be certified in the VANFLYTA REMS by enrolling and completing training.
- Prescribers must counsel patients receiving VANFLYTA about the risk of QT prolongation, torsades de pointes, and cardiac arrest, and provide patients with a Patient Wallet Card.
- Pharmacies that dispense VANFLYTA must be certified with the VANFLYTA REMS and must verify prescribers are certified through the VANFLYTA REMS.
Further information about the VANFLYTA REMS is available at www.VANFLYTAREMS.com or by telephone at 1-855-212-6670.
Embryo-Fetal Toxicity
Based on findings in animals and its mechanism of action, VANFLYTA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of quizartinib to pregnant rats during organogenesis at exposures 3 times the maximum recommended human dose (MRHD) of 53 mg/day caused structural abnormalities and alterations to growth.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with VANFLYTA and for 7 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with VANFLYTA and for 4 months after the last dose [see Use in Specific Populations (8.1 , 8.3) ] .
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- QT Prolongation, Torsades de Pointes, and Cardiac Arrest [see Warnings and Precautions (5.1) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Newly Diagnosed FLT3-ITD positive AML
The safety of VANFLYTA (35.4 mg orally once daily with chemotherapy, 26.5 mg to 53 mg orally once daily as maintenance) in adult patients with newly diagnosed FLT3-ITD positive AML is based on QuANTUM-First, a randomized, double-blind clinical trial of VANFLYTA (n=265) or placebo (n=268) with chemotherapy [see Clinical Studies (14) ] . Among patients who received VANFLYTA, 38% were exposed for 6 months or longer and 30% were exposed for greater than one year. On the VANFLYTA plus chemotherapy arm, 65% and 44% of patients completed induction and consolidation therapy, respectively, compared to 65% and 34% of patients in the placebo plus chemotherapy arm.
Serious adverse reactions in ≥5% of patients who received VANFLYTA plus chemotherapy were: febrile neutropenia (11%). Fatal adverse reactions occurred in 10% of patients who received VANFLYTA plus chemotherapy, including sepsis (5%), fungal infections (0.8%), brain edema (0.8%), and one case each of febrile neutropenia, pneumonia, cerebral infarction, acute respiratory distress syndrome, pulmonary embolism, ventricular dysfunction, and cardiac arrest.
Permanent discontinuation due to an adverse reaction in patients in the VANFLYTA plus chemotherapy arm occurred in 20% of patients. The most frequent (≥2%) adverse reaction which resulted in permanent discontinuation in the VANFLYTA arm was sepsis (5%).
Dosage interruptions of VANFLYTA due to an adverse reaction occurred in 34% of patients. Adverse reactions which required dosage interruption in ≥2% of patients in the VANFLYTA arm included neutropenia (11%), thrombocytopenia (5%), and myelosuppression (3%).
Dose reductions of VANFLYTA due to an adverse reaction occurred in 19% of patients. Adverse reactions which required dosage reductions in ≥2% of patients in the VANFLYTA arm were neutropenia (9%), thrombocytopenia (5%), and electrocardiogram QT prolonged (4%).
The most common adverse reactions (≥10% with a difference between arms of ≥2% compared to placebo), including laboratory abnormalities, were lymphocytes decreased, potassium decreased, albumin decreased, phosphorus decreased, alkaline phosphatase increased, magnesium decreased, febrile neutropenia, diarrhea, mucositis, nausea, calcium decreased, abdominal pain, sepsis, neutropenia, headache, creatine phosphokinase increased, vomiting, upper respiratory tract infections, hypertransaminasemia, thrombocytopenia, decreased appetite, fungal infections, epistaxis, potassium increased, herpesvirus infections, insomnia, electrocardiogram QT prolonged, magnesium increased, sodium increased, dyspepsia, anemia, and eye irritation.
Tables 5 and 6 summarize adverse reactions and laboratory abnormalities observed in patients receiving VANFLYTA in the clinical trial.
| Body System Adverse Reaction | VANFLYTA + Chemotherapy (N=265) | PLACEBO + Chemotherapy (N=268) | ||
|---|---|---|---|---|
| All Grades % | Grade 3 or 4 % | All Grades % | Grade 3 or 4 % | |
| Blood and Lymphatic System Disorders | ||||
| Febrile neutropenia Including fatalities. | 44 | 43 | 42 | 41 |
| Neutropenia Includes other related terms. | 29 | 26 | 14 | 12 |
| Thrombocytopenia | 18 | 13 | 13 | 12 |
| Anemia | 11 | 6 | 7 | 5 |
| Gastrointestinal Disorders | ||||
| Diarrhea Diarrhea includes colitis, diarrhea, enteritis, enterocolitis, gastroenteritis, and neutropenic colitis. | 42 | 8 | 39 | 8 |
| Mucositis Mucositis includes anal inflammation, anal ulcer, anorectal discomfort, aphthous ulcer, laryngeal inflammation, laryngeal pain, mucosal inflammation, edema mucosal, esophageal pain, esophageal ulcer, esophagitis, oral blood blister, oral disorder, oral mucosa erosion, oral mucosal blistering, oral mucosal erythema, oral pain, oropharyngeal pain, pharyngeal inflammation, proctalgia, proctitis, stomatitis, tongue ulceration, and vaginal ulceration. | 38 | 5 | 33 | 4.1 |
| Nausea | 34 | 1.5 | 31 | 1.9 |
| Abdominal pain | 30 | 2.3 | 22 | 1.1 |
| Vomiting | 25 | 0 | 20 | 1.5 |
| Dyspepsia | 11 | 0.4 | 9 | 0.7 |
| Infections and Infestations | ||||
| Sepsis Sepsis includes acinetobacter infection, bacteremia, bacterial sepsis, corynebacterium bacteremia, device related bacteremia, device related sepsis, enterobacter sepsis, enterococcal bacteremia, enterococcal sepsis, escherichia bacteremia, escherichia sepsis, klebsiella bacteremia, klebsiella sepsis, neutropenic sepsis, pseudomonal bacteremia, pulmonary sepsis, sepsis, septic shock, staphylococcal bacteremia, staphylococcal infection, staphylococcal sepsis, stenotrophomonas sepsis, streptococcal sepsis, and streptococcal bacteremia. , | 30 | 19 | 26 | 20 |
| Upper respiratory tract infection | 21 | 2.6 | 12 | 3 |
| Fungal infection Fungal infection includes aspergillosis oral, aspergillus infection, bronchopulmonary aspergillosis, candida infection, candida sepsis, fungal infection, fungal sepsis, fungal skin infection, fusarium infection, gastrointestinal candidiasis, hepatic infection fungal, hepatosplenic candidiasis, lower respiratory tract infection fungal, mucormycosis, oral candidiasis, oral fungal infection, oropharyngeal candidiasis, systemic candida, systemic mycosis, tinea cruris, and vulvovaginal candidiasis. , | 16 | 6 | 10 | 3 |
| Herpesvirus infection Herpesvirus infection includes disseminated varicella zoster virus infection, genital herpes, herpes simplex, herpesvirus infection, herpes zoster, oral herpes, and varicella zoster virus infection. | 14 | 2.6 | 8 | 1.9 |
| Nervous System Disorders | ||||
| Headache | 28 | 0 | 20 | 0.7 |
| Hepatobiliary disorders | ||||
| Hypertransaminasemia Hypertransaminasemia includes alanine aminotransferase increased, aspartate aminotransferase increased, transaminases increased, hepatic enzymes increased, and hypertransaminasemia. | 19 | 7 | 14 | 6 |
| Metabolism and Nutrition Disorders | ||||
| Decreased appetite | 17 | 4.9 | 13 | 1.9 |
| Respiratory, Thoracic and Mediastinal Disorders | ||||
| Epistaxis | 15 | 1.1 | 11 | 0.4 |
| Psychiatric Disorders | ||||
| Insomnia | 14 | 0 | 11 | 0 |
| Investigations | ||||
| Electrocardiogram QT prolonged | 14 | 3 | 4.1 | 1.1 |
| Eye Disorders | ||||
| Eye irritation Eye irritation includes dry eye, eye inflammation, eye irritation, eye pain, eye pruritus, foreign body sensation in eyes, keratitis, and ulcerative keratitis. | 11 | 0 | 7 | 0 |
Laboratory Abnormalities
Prolonged thrombocytopenia or neutropenia in the absence of active leukemia lasting past cycle day 42 of induction cycle 1 were noted in 8% of patients on the VANFLYTA plus chemotherapy arm and 4% of patients in the placebo plus chemotherapy arm.
| Laboratory Abnormality | VANFLYTA + Chemotherapy The denominator used to calculate the rate varied from 199 to 260 in VANFLYTA + Chemotherapy and from 187 to 267 in PLACEBO + Chemotherapy based on the number of patients with a baseline value and at least one post-treatment value. | PLACEBO + Chemotherapy | ||
|---|---|---|---|---|
| All Grades% | Grades 3 or 4% | All Grades% | Grades 3 or 4% | |
| Lymphocytes decreased | 60 | 57 | 55 | 51 |
| Potassium decreased | 59 | 22 | 56 | 18 |
| Albumin decreased | 53 | 1.6 | 45 | 4.3 |
| Phosphorus decreased | 52 | 22 | 48 | 19 |
| Alkaline phosphatase increased | 51 | 1.6 | 47 | 1.9 |
| Magnesium decreased | 44 | 2 | 42 | 1.1 |
| Calcium decreased | 33 | 2.4 | 27 | 1.6 |
| Creatine phosphokinase increased | 26 | 2.5 | 7 | 0.5 |
| Potassium increased | 15 | 1.2 | 11 | 0.8 |
| Magnesium increased | 14 | 2.8 | 9 | 1.2 |
| Sodium increased | 13 | 0 | 10 | 0.4 |
Other Clinical Trials
Clinically relevant adverse reactions in <10% of patients who received quizartinib for relapsed or refractory FLT3-ITD positive AML, an indication for which VANFLYTA is not approved, included differentiation syndrome (5%) and acute febrile neutrophilic dermatosis (3%).
DRUG INTERACTIONS
| Strong CYP3A Inhibitors | |
| Clinical Impact | VANFLYTA is a CYP3A substrate. Concomitant use of VANFLYTA with a strong CYP3A inhibitor increases quizartinib systemic exposure [see Clinical Pharmacology (12.3) ] , which may increase the risk of VANFLYTA adverse reactions. |
| Prevention or Management | Reduce the dosage of VANFLYTA [see Dosage and Administration (2.4) ]. |
| Strong or Moderate CYP3A Inducers | |
| Clinical Impact | Concomitant use of VANFLYTA with strong or moderate CYP3A inducers decreases quizartinib systemic exposure [see Clinical Pharmacology (12.3) ] , which may reduce VANFLYTA efficacy. |
| Prevention or Management | Avoid concomitant use of VANFLYTA with strong or moderate CYP3A inducers [see Clinical Pharmacology (12.3) ] . |
| QT Interval Prolonging Drugs | |
| Clinical Impact | VANFLYTA prolongs the QT/QTc interval. Coadministration of VANFLYTA with other drugs that prolong the QT interval may further increase the incidence of QT prolongation [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2) ] . |
| Prevention or Management | Monitor patients more frequently with ECG if co-administration of VANFLYTA with drugs known to prolong the QT interval is required. Examples of QT prolonging drugs include but are not limited to antifungal azoles, ondansetron, granisetron, azithromycin, pentamidine, doxycycline, moxifloxacin, atovaquone, prochlorperazine, and tacrolimus. |
| Breast Cancer Resistant Protein (BCRP) Substrates | |
| Clinical Impact | Concomitant use of VANFLYTA with BCRP substrates may increase the risk of BCRP substrate-associated adverse reactions [see Clinical Pharmacology (12.3) ] . |
| Prevention or Management | Avoid concomitant use of VANFLYTA with drugs that are BCRP substrates where possible. If concomitant use is unavoidable, monitor patients more frequently for BCRP substrate-associated adverse reactions and decrease the BCRP substrates dosage(s) in accordance with the respective Prescribing Information. |
DESCRIPTION
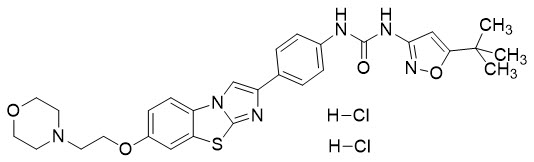
VANFLYTA (quizartinib) is a kinase inhibitor for oral use. The chemical name of quizartinib dihydrochloride is N -(5- tert -butyl-isoxazol-3-yl)- N '-{4-[7-(2-(morpholin-4-ylethoxy)imidazo[2,1- b ][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride. Quizartinib dihydrochloride is a white to off-white solid with a molecular formula of C 29 H 32 N 6 O 4 S∙2 HCl and a molecular weight of 633.6 for the salt and 560.7 for the free base. The aqueous solubility of quizartinib dihydrochloride (pKa 4.75 and 3.16) decreases with increasing pH. It is very slightly soluble in aqueous media at pH 1 and practically insoluble or insoluble at pH 2 and higher. Quizartinib dihydrochloride is very slightly soluble in ethanol. The chemical structure of quizartinib dihydrochloride is:
VANFLYTA is supplied as film-coated tablets containing 20 mg and 30 mg quizartinib dihydrochloride equivalent to 17.7 mg or 26.5 mg of quizartinib, respectively. The inactive ingredients in the tablet core are hydroxypropyl betadex, magnesium stearate, and microcrystalline cellulose. The tablet coating consists of hypromellose, talc, titanium dioxide, and triacetin. The 26.5 mg tablet coating also contains ferric oxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Quizartinib is a small molecule inhibitor of the receptor tyrosine kinase FLT3. Quizartinib and its major active metabolite AC886 bind to the adenosine triphosphate (ATP) binding domain of FLT3 with comparable affinity, and both had 10-fold lower affinity towards FLT3-ITD mutation compared to FLT3 in a binding assay. Quizartinib and AC886 inhibited FLT3 kinase activity, preventing autophosphorylation of the receptor, thereby inhibiting downstream FLT3 receptor signaling and blocking FLT3-ITD-dependent cell proliferation. Quizartinib showed antitumor activity in a mouse model of FLT3-ITD-dependent leukemia.
Pharmacodynamics
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of quizartinib have not been fully characterized.
Cardiac Electrophysiology
In vitro studies have shown that quizartinib is a predominant inhibitor of the slow delayed rectifier potassium current, I Ks .
The exposure-response analysis predicted a concentration-dependent QTcF interval median prolongation of 18 and 24 ms [upper bound of 2-sided 90% confidence interval (CI): 21 and 27 ms] at the median steady-state C max of quizartinib at the 26.5 mg and 53 mg dose level during maintenance therapy [see Warnings and Precautions (5.1) and Drug Interactions (7) ] .
Pharmacokinetics
The pharmacokinetics of quizartinib and its major circulating active metabolite (AC886) were characterized in healthy subjects and in patients with cancer and are presented as geometric mean (percent coefficient of variation [%CV]) unless otherwise specified.
Steady state quizartinib concentrations were achieved at Day 15 following once daily dosage in patients with AML. Quizartinib exposure (AUC and C max ) increased proportionally in the dose range of 26.5 mg (0.75 times the recommended 35.4 mg induction dose) to 79.5 mg (2.25 times the recommended 35.4 mg induction dose) in healthy subjects and 17.7 mg (0.5 times the recommended 35.4 mg induction dose) to 53 mg (1.5 times the recommended 35.4 mg induction dose) in patients with AML.
| Quizartinib | AC886 | |
|---|---|---|
| General Information | ||
| Steady state exposure following 35.4 mg VANFLYTA once daily during induction therapy in patients with newly diagnosed AML | ||
| C max | 140 ng/mL (71%) | 163 ng/mL (52%) |
| AUC 0-24h | 2,680 ng∙h/mL (85%) | 3,590 ng∙h/mL (51%) |
| Steady state exposure following 35.4 mg VANFLYTA once daily during consolidation therapy in patients with newly diagnosed AML | ||
| C max | 204 ng/mL (64%) | 172 ng/mL (47%) |
| AUC 0-24h | 3,930 ng∙h/mL (78%) | 3,800 ng∙h/mL (46%) |
| Steady state exposure following 53 mg VANFLYTA once daily during maintenance therapy in patients with newly diagnosed AML | ||
| C max | 529 ng/mL (60%) | 262 ng/mL (48%) |
| AUC 0-24h | 10,200 ng∙h/mL (75%) | 5,790 ng∙h/mL (46%) |
| Accumulation ratio Mean (±SD) (AUC 0-24h ) | 5.4 (4.4) | 8.7 (6.8) |
Absorption
The mean (SD) absolute bioavailability of quizartinib from the tablet formulation was 71% (±7%) in healthy subjects. After oral administration under fasted conditions, time to peak concentration (median T max ) of quizartinib and AC886 measured post dose was approximately 4 hours (range 2 to 8 hours) and 5 to 6 hours (range 4 to 120 hours), respectively, in healthy subjects.
No clinically significant differences in the pharmacokinetics of quizartinib were observed when administered with a high-fat, high-calorie meal.
Distribution
Volume of distribution at steady state in healthy subjects was estimated to be 275 L (17%).
In vitro plasma protein binding of quizartinib and AC886 is 99% or greater.
In vitro blood-to-plasma ratio for quizartinib and AC886 ranges from 0.79-1.30 and 1.36-3.19, respectively.
Elimination
Total body clearance of quizartinib in healthy subjects was estimated to be 2.23 L/hour (29%).
The mean (SD) effective half-lives (t 1/2 ) in patients with newly diagnosed AML for quizartinib and AC886 during maintenance therapy are 81 hours (±73) and 136 hours (±113), respectively.
Metabolism
In vitro quizartinib is primarily metabolized via oxidation by CYP3A4/5 and AC886 is formed and metabolized by CYP3A4/5.
Excretion
Following a single radiolabeled dose of quizartinib 53 mg to healthy subjects, 76.3% of the total radioactivity was recovered in feces (4% unchanged) and 1.64% in urine.
Specific Populations
There were no clinically significant differences in the exposure of quizartinib and AC886 based on age (range 18 to 91 years), sex, race (White 65%, Asian 18%, Black 9%), or body weight (range 37 to 153 kg).
Patients with Renal Impairment
Mild or moderate renal impairment (i.e., estimated creatinine clearance [CLcr] by Cockcroft-Gault equation: 30 to 89 mL/min) were not associated with clinically significant differences in the exposure of quizartinib and AC886.
The impact of severe renal impairment (CLcr <30 mL/min) on the pharmacokinetics of quizartinib and AC886 is unknown [see Use in Specific Populations (8.6) ] .
Patients with Hepatic Impairment
Mild (total bilirubin ≤ upper limit of normal [ULN] and aspartate aminotransferase [AST] >ULN or total bilirubin >1 to 1.5 times ULN and any value for AST) or moderate (total bilirubin >1.5 to 3 times ULN and any value for AST) hepatic impairment were not associated with clinically significant differences in the exposure of quizartinib and AC886.
The impact of severe hepatic impairment (Child-Pugh Class C or total bilirubin >3 times ULN and any value for AST) on the pharmacokinetics of quizartinib and AC886 is unknown [see Use in Specific Populations (8.7) ] .
Drug Interaction Studies and Model-Informed Approaches
Clinical Studies
Strong CYP3A Inhibitors
The AUC of quizartinib increased by 94% and the C max by 17% following coadministration of a single 26.5 mg quizartinib dose with ketoconazole (a strong CYP3A inhibitor). The AUCinf of AC886 decreased by 15% and C max by 60%.
Moderate CYP3A Inhibitors
The AUC of quizartinib increased by 20% and the C max by 11% following coadministration of a single 26.5 mg quizartinib dose with fluconazole (a moderate CYP3A inhibitor). The AUCinf of AC886 increased by 14% and C max by 2%.
Strong or Moderate CYP3A Inducers
The AUC inf of quizartinib decreased by 90% and C max by 45% following concomitant use of a single 53 mg dose of quizartinib with efavirenz (a moderate CYP3A inducer). The AUC of AC886 decreased by 96% and the C max by 68%. The effect of concomitant use with a strong CYP3A inducer may result in even greater effect on quizartinib pharmacokinetics based on mechanistic understanding of the drugs involved.
The AUC inf of quizartinib decreased by 34% and C max by 19.4% following concomitant use of a single 53 mg dose of quizartinib with rufinamide (a weak CYP3A inducer). The AUC inf of AC886 decreased by 51.9% and the C max by 46.9%.
Gastric Acid Reducing Agents
No clinically significant differences in quizartinib pharmacokinetics were observed when used concomitantly with lansoprazole (a proton pump inhibitor that reduces gastric acid).
Other Drugs
There were no clinically significant differences in the pharmacokinetics of P-gp substrates (dabigatran etexilate) or UGT1A1 substrates (raltegravir) when used concomitantly with quizartinib.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes
Quizartinib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4. Quizartinib does not induce CYP3A4, CYP1A2, or CYP2B6.
AC886 does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP2C19, or CYP3A4. AC886 does not induce CYP3A4, CYP1A2, or CYP2B6.
Other Metabolic Pathways
Quizartinib inhibits UGT1A1.
Transporters
Quizartinib is a substrate for P-gp but not for BCRP, OATP1B1, OATP1B3, OCT1, OAT2, MATE1, or MRP2. AC886 is a substrate for BCRP but not for OATP1B1, OATP1B3, MATE1, or MRP2.
Quizartinib is expected to inhibit BCRP at the recommended dosage.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with quizartinib.
Quizartinib was mutagenic in a bacterial reverse mutation (Ames) assay and not mutagenic in an in vivo transgenic rat mutation assay. Quizartinib was not genotoxic in vitro in mouse lymphoma thymidine kinase mutation and human lymphocyte chromosome aberration assays, or in an in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with quizartinib. However, adverse findings in male and female reproductive systems were observed in repeat-dose toxicity studies in rats and monkeys. Findings in female animals (rats or monkeys) included ovarian cysts, vaginal mucosal modifications, and atrophy of the uterus, ovary, and vagina, starting at exposures (AUC) approximately 0.2 times the MRHD of 53 mg/day. In male animals (rats and monkeys), findings included testicular seminiferous tubular degeneration, failure of sperm release, germ cell depletion in the testes, and oligospermia/aspermia, starting at exposures approximately 0.4 times the MRHD. After approximately one month of recovery period, all these findings except the vaginal mucosal modifications in the female rats were reversible.
CLINICAL STUDIES
The efficacy of VANFLYTA in combination with chemotherapy was evaluated in QuANTUM-First (NCT02668653), a randomized, double-blind, placebo-controlled study of 539 patients with newly diagnosed FLT3-ITD positive AML. FLT3-ITD status was determined prospectively with a clinical trial assay and verified retrospectively using the companion diagnostic LeukoStrat ® CDx FLT3 Mutation Assay, which is an FDA-approved test for selection of patients with AML for VANFLYTA treatment.
Patients were stratified by age (<60 versus ≥60 years), white blood cell count at diagnosis (<40×10 9 /L versus ≥40×10 9 /L), and region (North America, Europe versus Asia, other regions). Patients were randomized (1:1) to receive VANFLYTA (n=268) or placebo (n=271) in combination with induction and consolidation therapy and as maintenance monotherapy according to the initial assignment, as follows: a) Induction : 35.4 mg orally once daily on Days 8-21 of 7 + 3 (cytarabine [100 or 200 mg/m 2 /day] on Days 1 to 7 plus daunorubicin [60 mg/m 2 /day] or idarubicin [12 mg/m 2 /day] on Days 1 to 3) and on Days 8-21 or 6-19 of an optional second induction (7 + 3 or 5 + 2 [5 days cytarabine plus 2 days daunorubicin or idarubicin], respectively), b) Consolidation : 35.4 mg orally once daily on Days 6-19 of high-dose cytarabine (1.5 to 3 g/m 2 every 12 hours on Days 1, 3 and 5) for up to 4 cycles, and c) Maintenance : 26.5 mg orally once daily on Days 1 to 14 and 53 mg once daily thereafter for up to thirty-six 28-day cycles. There was no re-randomization at the start of post consolidation therapy. Patients who proceeded to hematopoietic stem cell transplantation (HSCT) initiated maintenance therapy after recovery from the HSCT.
The two treatment groups were generally balanced with respect to baseline demographics and disease characteristics. Of the 539 randomized patients, the median age was 56 years (range 20-75 years); 46% were male; 60% were White, 29% were Asian, 1% were Black, and 10% were other races. Eighty-four percent had an Eastern Cooperative Oncology Group (ECOG) baseline performance status of 0 or 1. The majority of the patients (72%) had intermediate risk cytogenetics at baseline. FLT3-ITD variant allelic frequency (VAF) was 3-25% in 36% of patients, >25-50% in 52% of patients, and >50% in 12% of patients. NPM1 mutations were identified in 52% of patients.
A second course of induction was administered to 20% of the patients, 65% initiated at least one cycle of consolidation, and 39% initiated maintenance. Among the patients who entered maintenance, 64% completed at least 12 cycles, 36% completed at least 24 cycles, and 16% completed all 36 planned cycles of maintenance. Twenty-nine percent (157/539) of the patients underwent HSCT in first complete remission (CR). The overall rate of HSCT (including the following settings: first CR, induction failure, or salvage after relapse) was 54% (144/268) in the VANFLYTA plus standard chemotherapy arm versus 47% (128/271) in the placebo plus standard chemotherapy arm. All patients were followed for survival.
Efficacy was established on the basis of overall survival (OS), measured from the date of randomization until death by any cause. The primary analysis was conducted after a minimum follow-up of 24 months after the randomization of the last patient. The study demonstrated a statistically significant improvement in OS for the VANFLYTA arm [hazard ratio (HR) 0.78; 95% CI: 0.62, 0.98; 2-sided p=0.0324] (see Figure 1 ).
| Figure 1: Kaplan-Meier Curve for Overall Survival in QuANTUM-First |
|---|
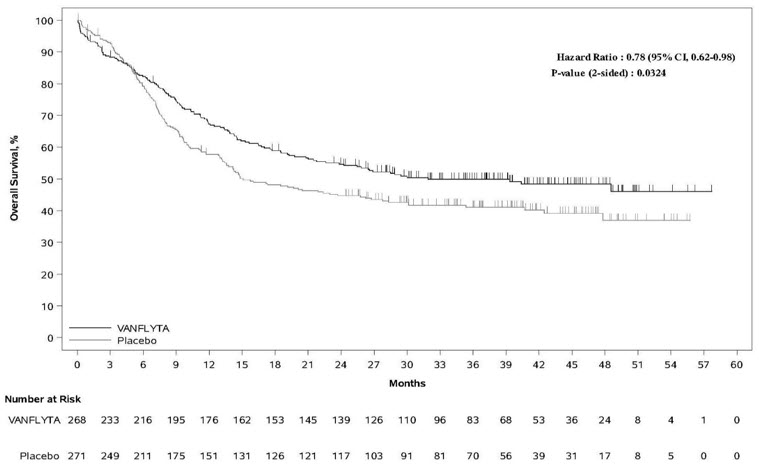 |
In an exploratory subgroup analysis of the 89/208 (43%) of patients who received maintenance therapy with VANFLYTA or placebo following consolidation chemotherapy, the OS HR was 0.40 (95% CI: 0.19, 0.84). Of 119/208 (57%) of patients who received maintenance therapy with VANFLYTA or placebo following HSCT, the OS HR was 1.62 (95% CI: 0.62, 4.22).
The CR rate in the VANFLYTA arm was 55% (95% CI: 48.7, 60.9) with a median duration of CR of 38.6 months (95% CI: 21.9, NE), and the CR rate in the placebo arm was 55% (95% CI: 49.2, 61.4) with a median duration of CR of 12.4 months (95% CI: 8.8, 22.7).
HOW SUPPLIED/STORAGE AND HANDLING
VANFLYTA (quizartinib) is supplied as round, film-coated tablets, packaged in bottles with a child-resistant closure.
Store at 20°C to 25°C (68°F to 77°F); excursions permitted from 15°C to 30°C (59°F to 86°F) [See USP Controlled Room Temperature].
| Tablet Strength | Tablet Description | Package Configuration | NDC |
|---|---|---|---|
| 17.7 mg | White, round, film-coated, debossed with "DSC511" , | 28-count bottle | 65597-504-28 |
| 14-count bottle | 65597-504-04 | ||
| 26.5 mg | Yellow, round, film-coated, debossed with "DSC512" | 28-count bottle | 65597-511-28 |
| 14-count bottle | 65597-511-04 |
Mechanism of Action
Quizartinib is a small molecule inhibitor of the receptor tyrosine kinase FLT3. Quizartinib and its major active metabolite AC886 bind to the adenosine triphosphate (ATP) binding domain of FLT3 with comparable affinity, and both had 10-fold lower affinity towards FLT3-ITD mutation compared to FLT3 in a binding assay. Quizartinib and AC886 inhibited FLT3 kinase activity, preventing autophosphorylation of the receptor, thereby inhibiting downstream FLT3 receptor signaling and blocking FLT3-ITD-dependent cell proliferation. Quizartinib showed antitumor activity in a mouse model of FLT3-ITD-dependent leukemia.