Get your patient on Xphozah (Tenapanor)
Patient education
Patient education materials
Treatment initiation and patient onboarding
Patient support program
Dosing resources
Clinical information
Insurance resources
Prior authorization & coverage support
Financial assistance & copay programs
Specialty pharmacy coordination
Legal resources
Other resources
Dosage & administration

Xphozah prescribing information
INDICATIONS AND USAGE
XPHOZAH is indicated to reduce serum phosphorus in adults with chronic kidney disease (CKD) on dialysis as add-on therapy in patients who have an inadequate response to phosphate binders or who are intolerant of any dose of phosphate binder therapy.
DOSAGE AND ADMINISTRATION
- Recommended dosage: 30 mg orally twice daily before the morning and evening meals (2.1 ).
- Manage serum phosphorus levels and tolerability with dosage adjustments (2.1 ).
- Take just prior to the first and last meals of the day (2.2 ).
- Instruct patients not to take right before a hemodialysis session, and instead take right before the next meal following dialysis (2.2 ).
Recommended Dosage
The recommended dosage is 30 mg orally twice daily before the morning and evening meals. Monitor serum phosphorus and adjust the dosage as needed to manage gastrointestinal tolerability.
Administration Instructions
- Instruct patients to take XPHOZAH just prior to the first and last meals of the day [see Clinical Pharmacology (12.2) ] .
- Instruct patients not to take XPHOZAH right before a hemodialysis session, and instead take right before the next meal following dialysis, as patients may experience diarrhea after taking XPHOZAH [see Warnings and Precautions (5.1) ] .
- Instruct patients who miss a dose to skip the missed dose and take the next dose at the regular time.
DOSAGE FORMS AND STRENGTHS
XPHOZAH tablets available as:
10 mg tenapanor supplied as a yellow, oval, biconvex film-coated tablet debossed with "
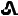 " on one side and "T10" on the other side.
" on one side and "T10" on the other side. 20 mg tenapanor supplied as a brown, oval, biconvex film-coated tablet debossed with "
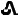 " on one side and "T20" on the other side.
" on one side and "T20" on the other side. 30 mg tenapanor supplied as a red, oval, biconvex film-coated tablet debossed with "
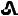 " on one side and "T30" on the other side.
" on one side and "T30" on the other side.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Tenapanor is essentially non-absorbed systemically, with plasma concentrations below the limit of quantification (less than 0.5 ng/mL) following oral administration [see Clinical Pharmacology (12.3) ]. Therefore, maternal use is not expected to result in fetal exposure to the drug.
The available data on XPHOZAH exposure from a small number of pregnant women have not identified any drug associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. In reproduction studies with tenapanor in pregnant rats and rabbits, no adverse fetal effects were observed in rats at 0.2 times the maximum recommended human dose and in rabbits at doses up to 15 times the maximum recommended human dose (based on body surface area) [see Nonclinical Toxicology (13.1) ] .
The estimated background risk of major birth defects and miscarriage for women with CKD on dialysis with hyperphosphatemia is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the United States general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Animal Data
In an embryofetal development study in rats, tenapanor was administered orally to pregnant rats during the period of organogenesis at dose levels of 1, 10 and 30 mg/kg/day. Tenapanor doses of 10 and 30 mg/kg/day were not tolerated by the pregnant rats and was associated with mortality and moribundity with body weight loss. The 10 and 30 mg/kg dose group animals were sacrificed early, and the fetuses were not examined for intrauterine parameters and fetal morphology. No adverse fetal effects were observed in rats at 1 mg/kg/day (approximately 0.2 times the maximum recommended human dose) and in rabbits at doses up to 45 mg/kg/day (approximately 15 times the maximum recommended human dose, based on body surface area). In a pre- and post-natal developmental study in mice, tenapanor at doses up to 200 mg/kg/day (approximately 16.5 times the maximum recommended human dose, based on body surface area) had no effect on pre- and post-natal development.
Lactation
Risk Summary
Tenapanor and its major metabolite, M1, were not detected in the breast milk of lactating women ( see Data ). In adults, concentrations of tenapanor were below the limit of quantitation in plasma following multiple doses of tenapanor [see Clinical Pharmacology (12.3) ]. Maternal use of XPHOZAH is not expected to result in exposure to tenapanor or its major metabolite in breastfed infants. There is no information on the effects of tenapanor or M1 on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for XPHOZAH and any potential adverse effects on the breastfed infant from XPHOZAH or from the underlying maternal condition.
Data
A clinical lactation study was conducted in seven healthy adult women who were 22 to 37 years of age. Following oral administration of tenapanor 50 mg twice daily for 3 days, the concentrations of tenapanor and its major metabolite were below the limit of quantitation (<1 ng/mL and <1 ng/mL) in all breast milk samples collected over 24 hours post dosing.
Pediatric Use
Risk Summary
XPHOZAH is contraindicated in patients less than 6 years of age. In nonclinical studies, deaths occurred in young juvenile rats (less than 1-week old rats; approximate human age-equivalent of less than 2 years of age) and in older juvenile rats (approximate human age-equivalent of 2 years of age) following oral administration of tenapanor, as described below in Juvenile Animal Toxicity Data.
The safety and effectiveness of XPHOZAH in pediatric patients have not been established.
Juvenile Animal Toxicity Data
In a 21-day oral dose range finding toxicity study in juvenile rats, tenapanor was administered to neonatal rats (post-natal day (PND) 5) at doses of 5 and 10 mg/kg/day. Tenapanor was not tolerated in male and female pups and the study was terminated on PND 16 due to mortalities and decreased body weight (24% to 29% reduction in females at the respective dose groups and 33% reduction in males in the 10 mg/kg/day group, compared to control).
In a second dose range finding study, tenapanor doses of 0.1, 0.5, 2.5, or 5 mg/kg/day were administered to neonatal rats from PND 5 through PND 24. Treatment-related mortalities were observed at 0.5, 2.5, and 5 mg/kg/day doses. These premature deaths were observed as early as PND 8, with majority of deaths occurring between PND 15 and 25. In the 5 mg/kg/day group, mean body weights were 47% lower for males on PND 23 and 35% lower for females on PND 22 when compared to the controls. Slightly lower mean tibial lengths (5% to 11%) were noted in males and females in the 0.5, 2.5, and 5 mg/kg/day dose groups on PND 25 and correlated with the decrements in body weight noted in these groups. Lower spleen, thymus, and/or ovarian weights were noted at the 0.5, 2.5, and 5 mg/kg/day doses. Tenapanor-related gastrointestinal distension and microscopic bone findings of increased osteoclasts, eroded bone, and/or decreased bone in sternum and/or femorotibial joint were noted in males and females in the 0.5, 2.5, and 5 mg/kg/day dose groups.
In juvenile rats administered tenapanor at 0.03, 0.1, or 0.3 mg/kg/day on PND 5 through PND 61, treatment-related mortalities were observed at 0.3 mg/kg/day. Lower mean body weight gains were noted in the 0.3 mg/kg/day group males and females compared to the control group primarily during PND 12–24 but continuing sporadically during the remainder of the dosing period; corresponding lower mean food consumption was noted in this group during PND 21–33. As a result, mean body weights were up to 15.8% and 16.8% lower in males and females, respectively, compared to the control group; the greatest difference was on PND 24 for males and PND 21 for females. Mean body weight in the 0.3 mg/kg/day group males was only 3.9% lower than the control group on PND 61. There were no tenapanor-related effects on mean body weights, body weight gains, or food consumption in the 0.03 and 0.1 mg/kg/day group males and females. A dosage level of 0.1 mg/kg/day was considered to be the no-observed-adverse-effect level (NOAEL) for juvenile toxicity of tenapanor [see Contraindications (4) , Warnings and Precautions (5.1) ].
In a 21-day oral dose range finding study in older (weaned) juvenile rats administered tenapanor at 0.1, 1, or 5 mg/kg/day on PND 21 through PND 41 (approximate human age-equivalent of 2 to 12 years of age), treatment-related mortalities or moribundities were observed during the first two days of the study in the 1 mg/kg/day males and the 5 mg/kg/day males and females. Watery feces, decreased food consumption, and lower mean body weight were also observed in the 1 and 5 mg/kg/day groups.
In weaned juvenile rats administered tenapanor at 0.1, 0.3, and 0.7 (males) or 1 (females) mg/kg/day on PND 21 through PND 80, no mortalities were observed. Significant decreases in mean body weights were observed in the 0.3 and 0.7 mg/kg/day males throughout the dosing period (up to 20.3% lower than control) and in the 1 mg/kg/day females between PND 23 to 35 (up to 16.7% lower than control), with food consumption notably decreased on PND 21 to 29. There were also reductions in tibia length between PND 76 and 80 in the 0.3 and 0.7 mg/kg/day males, and between PND 36 and 64 in the 0.7 mg/kg/day males, which were not observed during the 14-day recovery period. The NOAEL was considered to be 0.1 mg/kg/day for juvenile toxicity of tenapanor.
Geriatric Use
Of 1010 adult patients with CKD on dialysis randomized and treated in two randomized, double-blind, placebo-controlled randomized withdrawal clinical trials for XPHOZAH (TEN-02-201 and TEN-02-301) as well as a third randomized, double-blind, placebo-controlled trial (TEN-02-202) for XPHOZAH in combination with phosphate binders, 282 (28%) were 65 years of age and older. Clinical studies of XPHOZAH did not include sufficient numbers of patients aged 65 and older to determine whether they respond differently than younger patients.
CONTRAINDICATIONS
XPHOZAH is contraindicated in patients under 6 years of age because of the risk of diarrhea and serious dehydration [see Warnings and Precautions (5.1) , Use in Specific Populations (8.5) ].
XPHOZAH is contraindicated in patients with known or suspected mechanical gastrointestinal obstruction.
WARNINGS AND PRECAUTIONS
Patients may experience severe diarrhea (5.1 ).
Diarrhea
Diarrhea was the most common adverse reaction in XPHOZAH-treated patients with CKD on dialysis [see Dosage and Administration (2) , Contraindications (4) and Adverse Reactions (6.1) ]. In clinical trials, diarrhea was reported in up to 53% of patients, reported as severe in 5%, and associated with dehydration and hyponatremia in less than 1% of patients. Treatment with XPHOZAH should be discontinued in patients who develop severe diarrhea.
ADVERSE REACTIONS
Most common adverse reaction in the combined clinical trials was diarrhea, reported by 43-53% of patients (6 ).
To report SUSPECTED ADVERSE REACTIONS, contact Ardelyx at 1-844-974-6924 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described below reflect data from 754 adults with CKD on dialysis taking XPHOZAH in clinical trials as monotherapy and in combination with phosphate binders. Among the 754 patients, 258 patients were exposed to tenapanor for at least 26 weeks and 75 were exposed to tenapanor for at least one year. [see Clinical Studies (14) ] .
Most Common Adverse Reaction
Diarrhea, which occurred in 43-53% of patients, was the only adverse reaction reported in at least 5% of XPHOZAH-treated patients with CKD on dialysis across trials. The majority of diarrhea events in the XPHOZAH-treated patients were reported to be mild-to-moderate in severity and resolved over time, or with dose reduction. Diarrhea was typically reported soon after initiation but could occur at any time during treatment with XPHOZAH. Severe diarrhea was reported in 5% of XPHOZAH-treated patients in these trials [see Warnings and Precautions (5.1) ] .
Postmarketing Experience
The following adverse reactions have been identified during post approval use of XPHOZAH. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity reactions: pruritis, rash, and urticaria
DRUG INTERACTIONS
- OATP2B1 Substrates: Potential for reduced exposure of the concomitant drug (e.g., enalapril). Monitor for signs related to loss of efficacy and adjust the dosage of the concomitantly administered drug as needed (7.1 ).
- Sodium Polystyrene Sulfonate (SPS): Separate administration by at least three hours (7.2 ).
OATP2B1 Substrates
Tenapanor is an inhibitor of intestinal uptake transporter, OATP2B1 [see Clinical Pharmacology (12.3) ] . Drugs which are substrates of OATP2B1 may have reduced exposures when concomitantly taken with XPHOZAH. Monitor for signs related to loss of efficacy and adjust the dose of concomitantly administered drug as needed.
Enalapril is a substrate of OATP2B1. When enalapril was coadministered with XPHOZAH (30 mg twice daily for five days), the peak exposure (C max ) of enalapril and its active metabolite, enalaprilat, decreased by approximately 70% and total systemic exposures (AUC) decreased by 50 to 65% compared to when enalapril was administered alone [see Clinical Pharmacology (12.3) ] . However, the decrease in enalaprilat's exposure with XPHOZAH may be offset by the inherently higher exposures observed in patients with CKD on dialysis due to its reduced renal clearance. Therefore, a lower starting dose of enalapril, which is otherwise recommended in patients with CKD on dialysis is not required when enalapril is coadministered with XPHOZAH.
Sodium Polystyrene Sulfonate
Separate administration of XPHOZAH and sodium polystyrene sulfonate (SPS) by at least 3 hours. SPS binds to many commonly prescribed oral medicines.
DESCRIPTION
XPHOZAH (tenapanor) tablets contain tenapanor hydrochloride as an active ingredient. Tenapanor hydrochloride is a sodium/hydrogen exchanger 3 (NHE3) inhibitor for oral use. The chemical name for tenapanor hydrochloride is 12,15-Dioxa-2,7,9-triazaheptadecanamide, 17-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-, hydrochloride (1:2). Tenapanor hydrochloride has the molecular formula of C 50 H 68 Cl 6 N 8 O 10 S 2 , the molecular weight of 1218 Daltons, and the chemical structure below:
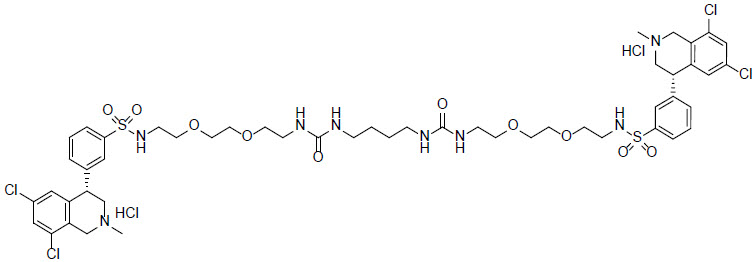
Tenapanor hydrochloride is a white to off-white to light brown hygroscopic amorphous solid. It is practically insoluble in water.
XPHOZAH tablets contain the equivalent of 10, 20, or 30 mg of tenapanor (provided as 10.6, 21.3, and 31.9 mg of tenapanor hydrochloride, respectively). Inactive ingredients in the tablet are colloidal silicon dioxide, low-substituted hydroxypropyl cellulose, microcrystalline cellulose, propyl gallate, stearic acid, tartaric acid, and the coating agent OPADRY ® , which consists of hypromellose, titanium dioxide and triacetin, as well as iron oxide yellow as colorant [10 mg and 20 mg tablets], iron oxide red as colorant [20 mg and 30 mg tablets] and iron oxide black as colorant [30 mg tablet].
CLINICAL PHARMACOLOGY
Mechanism of Action
Tenapanor is a locally acting inhibitor that targets the sodium/hydrogen exchanger 3 (NHE3), an antiporter expressed on the apical surface of the epithelium of the small intestine and colon. Inhibition of NHE3 by tenapanor results in reduced sodium absorption and decreased phosphate absorption by reducing phosphate permeability through the paracellular pathway.
Pharmacodynamics
Cardiac Electrophysiology
At 3 times the mean maximum exposure of the major pharmacologically inactive metabolite of tenapanor (M1) at the recommended dosage, there were no clinically relevant effects on the QTc.
Food Effect
Administration of XPHOZAH 5 to 10 minutes before a meal increased the 24-hour stool phosphorus excretion compared to taking XPHOZAH in the fed or fasting condition [see Dosage and Administration (2.2) ] . In clinical trials, XPHOZAH was administered just prior to the first meal and just prior to dinner.
Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of XPHOZAH was studied in 20 subjects with normal or moderate hepatic impairment function (Child-Pugh B) after a single 100-mg dose. Compared to subjects with normal hepatic function, the geometric mean C max of the pharmacologically inactive metabolite M1, was approximately 27% to 35% lower in subjects with moderate hepatic impairment. Tenapanor exhibited minimal systemic bioavailability in subjects with both normal hepatic function and moderate hepatic impairment, with plasma concentrations below the limit of quantitation (less than 0.5 ng/mL) in the majority of samples.
Pharmacokinetics
Absorption
Tenapanor is minimally absorbed following repeated twice daily oral administration. Plasma concentrations of tenapanor were below the limit of quantitation (less than 0.5 ng/mL) in the majority of samples from subjects following single and repeated oral administration of tenapanor 30 mg twice daily. Therefore, standard pharmacokinetic parameters such as area under the curve (AUC), maximum concentration (C max ), and half-life (t 1/2 ) could not be determined.
Distribution
Plasma protein binding of tenapanor and its major metabolite, M1, is approximately 99% and 97%, respectively , in vitro.
Elimination
Metabolism
Tenapanor is metabolized primarily by CYP3A4/5 and low levels of its major metabolite, M1, are detected in plasma. The C max of M1 is approximately 3 ng/mL after a single dose of XPHOZAH 30 mg and 14 ng/mL at steady state following repeated dosing of XPHOZAH 30 mg twice daily in healthy subjects. Based on a cross-study comparison, the steady state plasma concentration of M1 in CKD patients on dialysis (eGFR less than 15 mL/min/1.73 m 2 ) was not notably different from those of healthy subjects given similar doses of XPHOZAH.
Excretion
Following administration of a single 15 mg radiolabeled 14 C- XPHOZAH dose to healthy subjects, approximately 70% of the radioactivity was excreted in feces through 120 hours post-dose (79% through 240 hours post-dose), mostly as the parent drug accounting for 65% of dose within 144 hours post-dose. Approximately 9% of the administered dose was recovered in urine, primarily as metabolites. M1 is excreted in urine unchanged accounting for 1.5% of dose within 144 hours post-dose.
Drug Interaction Studies
CYP Metabolism Mediated Drug Interactions
Tenapanor and M1 did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6 in vitro .
Tenapanor and M1 did not induce CYP1A2 and CYP2B6 in vitro.
No significant inhibition or induction of CYP3A4 enzyme using midazolam as a substrate was observed when XPHOZAH 50 mg was administered twice a day for 13 days in healthy subjects.
No significant effect on PepT1 activity using cefadroxil as a substrate was observed when tenapanor 50 mg was administered twice a day for 12 days in healthy subjects.
No significant effect on P-gp and CYP2C9 activity using digoxin and warfarin as a substrate was observed when tenapanor 30 mg was administered twice a day for 12 days in healthy subjects.
Following co-administration of a single dose of XPHOZAH 50 mg with repeated doses of itraconazole 200 mg, a CYP3A4 inhibitor, the mean AUC and C max of M1 was decreased 50% in healthy subjects. Plasma concentrations of tenapanor were mostly below the limit of quantitation (less than 0.5 ng/mL) after co-administration of itraconazole.
Following administration of a single 20 mg enalapril dose with tenapanor (30 mg BID) at steady state, the mean AUC and C max of enalapril was decreased by 64% and 69%, respectively, in healthy subjects. The mean AUC and C max of enalaprilat was decreased by 52% and 68%, respectively, in healthy subjects.
Membrane Transporter Mediated Drug Interactions
Tenapanor inhibited OATP2B1, but is not an inhibitor of P-gp, BCRP, OATP1B1, OATP1B3, and PEPT1. M1 did not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2-K.
Tenapanor is not a substrate of P-gp, BCRP, OATP1B1, and OATP1B3. M1 is a substrate of P-gp .
Interactions with Phosphate Binders
In vitro studies indicated the potential for tenapanor to bind to sevelamer carbonate but did not suggest the potential to bind to calcium carbonate or calcium acetate. In a clinical study, there did not appear to be a pharmacodynamic interaction between sevelamer carbonate (800 mg TID) and tenapanor (14 mg BID).
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of tenapanor was assessed in a 6-month carcinogenicity study in Tg rasH2 mice and in a 2-year carcinogenicity study in rats. Tenapanor was not tumorigenic at oral doses up to 100 mg/kg/day (approximately 7.5 times the recommended human dose, based on the body surface area) in male mice and 800 mg/kg/day (approximately 65 times the maximum recommended human dose, based on the body surface area) for female mice. Tenapanor was not tumorigenic in male and female rats at oral doses up to 5 mg/kg/day (approximately 0.8 times the recommended human dose, based on the body surface area). The major metabolite of tenapanor, M1, was not tumorigenic in Tg rasH2 mice at oral doses up to 165 mg/kg/day (approximately 13.3 times the maximum recommended human dose, based on the body surface area).
Mutagenesis
Tenapanor was not genotoxic in the in vitro bacterial reverse mutation (Ames) assays, an in vitro chromosomal aberration assay in cultured human peripheral blood lymphocytes or the in vivo micronucleus assays in mice and rats. The M1 metabolite of tenapanor was not genotoxic in the Ames assay and in an in vitro micronucleus assay using L5178Y cells.
Impairment of Fertility
Tenapanor had no effect on fertility or reproductive function in male rats at oral doses up to 10 mg/kg/day (approximately 1.6 times the recommended human dose, based on the body surface area) and in female mice at oral doses up to 50 mg/kg/day (approximately 4 times the recommended human dose, based on the body surface area).
CLINICAL STUDIES
The ability of XPHOZAH to lower serum phosphorus in adults with CKD on dialysis was evaluated in 3 trials: TEN-02-201 [NCT02675998], TEN-02-301 [NCT03427125]), and TEN-02-202 [NCT03824587]). Across these trials, the mean age of XPHOZAH-treated patients was 56 (range 24 to 88 years), 61% were males, 44% were White, 49% were Black/African American, 3% were Asian, 3% were American Indian or Alaska Native, and 1% were other.
Both monotherapy trials (TEN-02-201 and TEN-02-301) enrolled patients who, following a 3-week washout period, had an increase in serum phosphorus of at least 1.5 mg/dL (compared to pre-wash out value) and a serum phosphorus level of at least 6.0 mg/dL and not more than 10.0 mg/dL.
Study TEN-02-301
Study TEN-02-301 included a 26-week randomized, active-controlled open-label treatment period, followed by a 12-week, blinded placebo-controlled randomized withdrawal period. A total of 564 patients were randomized into the 26-week treatment period (423 to XPHOZAH and 141 to the control arm which was intended to provide controlled safety data). Among the 423 patients randomized to XPHOZAH, 255 patients (60%) completed the 26-week treatment period and were rerandomized 1:1 to remain on XPHOZAH (n=128) or receive placebo (n=127). During the randomized withdrawal phase, the phosphorus concentration rose in the placebo group by 0.7 mg/dL (95% CI: (0.2, 1.1), p=0.002) relative to patients who remained on XPHOZAH.
Study TEN-02-201
Study TEN-02-201 included an 8-week randomized, double-blind period that evaluated three dosing regimens of XPHOZAH (3 mg twice daily, 10 mg twice daily, or a titration regimen). This period was followed by a 4-week placebo-controlled randomized-withdrawal phase, during which patients were rerandomized 1:1 to their current XPHOZAH treatment or to placebo. Of the 219 patients included in the trial, 164 patients (75%) completed the 8-week randomized treatment period and were rerandomized 1:1 to receive XPHOZAH (n=82) or placebo (n=82). During the randomized withdrawal phase, the phosphorus concentration rose in the placebo group by 0.7 mg/dL (95% CI: (0.3, 1.2), p=0.003) relative to patients who remained on XPHOZAH.
Study TEN-02-202
Study TEN-02-202 was a randomized, parallel-group, double-blind, placebo-controlled study that evaluated the effect of XPHOZAH on the change in serum phosphorus when used as add-on therapy in patients on stable phosphate-binder therapy with serum phosphorus greater than or equal to 5.5 mg/dL. A total of 236 patients were randomized to receive XPHOZAH (n=117) or placebo BID (n=119) for 4 weeks. During the 4-week period, the serum phosphorus decreased by 0.7 mg/dL (95% CI: (0.3, 1.0), p=0.0004) in the add-on XPHOZAH group as compared to the add-on placebo group.
HOW SUPPLIED/STORAGE AND HANDLING
XPHOZAH is supplied as 10-, 20-, or 30-mg oval, biconvex, film-coated tablets supplied in white, opaque, high-density polyethylene bottles containing 60 or 14 tablets with a silica gel canister (as the desiccant) and screw-top polypropylene child-resistant cap lined with an induction-activated aluminum foil liner:
| Strength | Color | Deboss | NDC | |
|---|---|---|---|---|
| Bottle/14 | Bottle/60 | |||
| 10 mg | Yellow |
| 73154-110-14 | 73154-110-60 |
| 20 mg | Brown |
| 73154-120-14 | 73154-120-60 |
| 30 mg | Red |
| 73154-130-14 | 73154-130-60 |
 /T10
/T10  /T20
/T20  /T30
/T30 Storage
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature] . Keep bottle tightly closed to protect from moisture. Store and dispense in original bottle with the desiccant.
Mechanism of Action
Tenapanor is a locally acting inhibitor that targets the sodium/hydrogen exchanger 3 (NHE3), an antiporter expressed on the apical surface of the epithelium of the small intestine and colon. Inhibition of NHE3 by tenapanor results in reduced sodium absorption and decreased phosphate absorption by reducing phosphate permeability through the paracellular pathway.