Get your patient on Ztalmy (Ganaxolone)
Ztalmy patient education
Patient toolkit
Dosage & administration

Ztalmy prescribing information
INDICATIONS AND USAGE
ZTALMY is indicated for the treatment of seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) in patients 2 years of age and older.
DOSAGE AND ADMINISTRATION
- Administer ZTALMY orally three times daily with food. (2.1 )
- Titrate ZTALMY gradually according to the recommended schedules. See full prescribing information. (2.1 )
- Dosage for patients weighing 28 kg or less (2.1 ):
- the starting dosage is 2 mg/kg three times daily (6 mg/kg/day)
- the maximum dosage is 21 mg/kg three times daily (63 mg/kg/day).
- Dosage for patients weighing over 28 kg (2.1 ):
- the starting dosage is 50 mg three times daily (150 mg daily)
- the maximum dosage is 600 mg three times daily (1800 mg daily).
- Patients with severe hepatic impairment: see full prescribing information for dosage recommendation. (2.3 )
Dosage Information
ZTALMY is administered by mouth three times daily and must be taken with food [see Clinical Pharmacology (12.3) ] .
The recommended titration schedule and maintenance dosage are based on body weight for patients weighing 28 kg or less. Dosage recommendations for patients weighing 28 kg or less are included in Table 1, and dosage recommendations for patients weighing more than 28 kg are included in Table 2. Dosage should be increased based on tolerability no more frequently than every 7 days. Titration increments should not exceed those shown in Table 1 and Table 2.
| Dosage | Total Daily Dose | Day |
|---|---|---|
| 2 mg/kg three times daily | 6 mg/kg/day | 1 to 7 |
| 4 mg/kg three times daily | 12 mg/kg/day | 8 to 14 |
| 8 mg/kg three times daily | 24 mg/kg/day | 15 to 21 |
| 14 mg/kg three times daily | 42 mg/kg/day | 22 to 28 |
| 21 mg/kg three times daily | 63 mg/kg/day | 29 and thereafter |
| Dosage | Total Daily Dose | Day |
|---|---|---|
| 50 mg three times daily | 150 mg | 1 to 7 |
| 100 mg three times daily | 300 mg | 8 to 14 |
| 200 mg three times daily | 600 mg | 15 to 21 |
| 400 mg three times daily | 1,200 mg | 22 to 28 |
| 600 mg three times daily | 1,800 mg | 29 and thereafter |
Administration Instructions
See the Instructions for Use for complete instructions on how to properly prepare and administer ZTALMY.
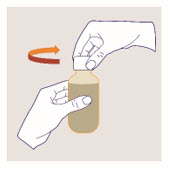
Shake the bottle thoroughly for at least 1 minute and then wait for 1 minute before measuring and administering each dose.
Measure and administer the prescribed dose using the oral syringe(s) provided by your pharmacist. A household teaspoon or tablespoon is not an adequate measuring device and should not be used.
ZTALMY must be administered with food [see Clinical Pharmacology (12.3) ] .
Discard any unused ZTALMY oral suspension after 30 days of first opening the bottle [see How Supplied/Storage and Handling (16.2) ] .
Dosage in Patients with Severe Hepatic Impairment
ZTALMY is administered by mouth three times daily and must be taken with food [see Clinical Pharmacology (12.3) ] .
The recommended titration schedule and maintenance dosage are based on body weight for patients weighing 28 kg or less. Dosage recommendations for patients with severe hepatic impairment and weighing 28 kg or less are included in Table 3, and dosage recommendations for patients with severe hepatic impairment (Child-Pugh class C) weighing more than 28 kg are included in Table 4. Dosage should be increased based on tolerability no more frequently than every 7 days. Titration increments should not exceed those shown in Table 3 and Table 4.
No dosage adjustment is necessary in patients with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment.
| Dosage | Total Daily Dose Approximate daily dose. | Days |
|---|---|---|
| 0.66 mg/kg three times daily | 2 mg/kg/day | 1 to 7 |
| 1.33 mg/kg three times daily | 4 mg/kg/day | 8 to 14 |
| 2.66 mg/kg three times daily | 8 mg/kg/day | 15 to 21 |
| 4.66 mg/kg three times daily | 14 mg/kg/day | 22 to 28 |
| 7 mg/kg three times daily | 21 mg/kg/day | 29 and thereafter |
| Dosage | Total Daily Dose Approximate daily dose. | Days |
|---|---|---|
| 17 mg three times daily | 50 mg | 1 to 7 |
| 33 mg three times daily | 100 mg | 8 to 14 |
| 67 mg three times daily | 200 mg | 15 to 21 |
| 133 mg three times daily | 400 mg | 22 to 28 |
| 200 mg three times daily | 600 mg | 29 and thereafter |
Discontinuation of ZTALMY
Decrease the dose of ZTALMY gradually when discontinuing treatment. As with all antiepileptic drugs, abrupt discontinuation should be avoided, when possible, to minimize the risk of increased seizure frequency and status epilepticus [see Warnings and Precautions (5.3) ] .
DOSAGE FORMS AND STRENGTHS
Oral suspension: 50 mg/mL ganaxolone. Each bottle contains 110 mL of white to off-white cherry flavored suspension.
USE IN SPECIFIC POPULATIONS
- Pregnancy: Based on animal data, may cause fetal harm. (8.1 )
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antiepileptic drugs (AEDs), such as ZTALMY, during pregnancy. Encourage women who are taking ZTALMY during pregnancy to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://www.aedpregnancyregistry.org/.
Risk Summary
There are no available data on ZTALMY use in pregnant women to inform a drug-associated risk of adverse developmental outcomes. In animal studies, adverse effects on development were observed in mice (fetal malformations) and rats (neurobehavioral and growth impairment) following exposure during organogenesis (mouse) or throughout gestation and lactation (rat) at maternal exposures lower than that in human adults at the maximum recommended human dose (MRHD) of 1800 mg. In addition, neuronal death was observed in rats exposed to ganaxolone during a period of brain development that begins during the third trimester of pregnancy in humans and continues during the first few years after birth.
In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risks of major birth defects and miscarriage for the indicated populations are unknown.
Epilepsy, with or without exposure to antiepileptic drugs, has been associated with several adverse outcomes during pregnancy, including preeclampsia, preterm labor, antepartum and postpartum hemorrhage, placental abruption, poor fetal growth, prematurity, fetal death, and maternal mortality. The risk of maternal or fetal injury may be greatest for patients with untreated or poorly controlled convulsive seizures. Women with epilepsy who become pregnant should not abruptly discontinue antiepileptic drugs, including ZTALMY, due to the risk of status epilepticus or severe seizures, which may be life-threatening [see Warnings and Precautions (5.3) ].
Data
Animal Data
In an embryofetal development study in mice, oral administration of ganaxolone (0, 50, 175, or 300 mg/kg/day) throughout the period of organogenesis resulted in increased incidences of fetal malformations (external and/or visceral) at all doses in the absence of maternal toxicity. Maternal plasma drug exposures (AUC) at the low-effect dose (50 mg/kg/day) for embryofetal developmental toxicity in the mouse were approximately 10-fold lower than that in humans at the MRHD.
In a combined embryofetal development and pre- and postnatal development study in rats, ganaxolone (0, 10, 20, or 40 mg/kg/day) was administered orally to females throughout gestation and lactation. There were no effects on embryofetal growth, survival, or morphology; however, adverse effects on offspring growth (delayed reflex development, decreased body weight gain) were observed during the postnatal period (prior to and after weaning) at the high dose, and neurobehavioral impairment (decreased locomotor activity) was observed in the offspring at the two highest doses. The no-effect dose (10 mg/kg/day) for pre- and postnatal developmental toxicity in rats was associated with maternal drug exposures less than that in humans at the MRHD.
Oral administration of ganaxolone (0, 20, 45, or 90 mg/kg/day) to rats on postnatal day (PND) 7 resulted in widespread apoptotic neurodegeneration in the brain (cortex, thalamus, and hippocampus) at all doses; a no-effect dose was not identified. Brain development on PND 7 in rat corresponds to that beginning in humans during the third trimester of pregnancy and continuing for the first several months to years after birth [see Use in Specific Populations (8.4) ] .
Lactation
Risk Summary
Ganaxolone is excreted in human milk. Following a single oral dose of ganaxolone (300 mg), ganaxolone exposures (AUC (0-24 h) ) in human milk were approximately 4 times higher than those in maternal plasma, resulting in an estimated daily dose in the infant of less than 1% of the maternal dose (see Data ) . The effects of ganaxolone on milk production and the breastfed infant are unknown.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZTALMY, and any potential adverse effects on the breastfed child from ZTALMY, or from the underlying maternal condition.
Data
A study was conducted in 5 healthy adult lactating women treated with a 300 mg oral dose of ganaxolone. Ganaxolone exposures in breast milk were approximately 4 times those in maternal plasma. The calculated maximum relative infant dose for ganaxolone is approximately 0.157 mg/kg/day based on an average milk intake of 150 mL/kg/day, which is less than 1% of the maternal dose, and approximately 0.24% the labeled pediatric dose of 63 mg/kg/day.
Pediatric Use
The safety and effectiveness of ZTALMY for the treatment of seizures associated with CDD have been established in pediatric patients 2 years of age and older.
The use of ZTALMY for the treatment of seizures associated with CDD in patients 2 years of age and older is supported by a randomized, double-blind, placebo-controlled trial that included 99 pediatric patients 2 to less than 18 years of age [see Clinical Studies (14) ] .
Safety and effectiveness of ZTALMY in pediatric patients below 2 years of age have not been established.
Juvenile Animal Data
Oral administration of ganaxolone (0, 20, 45, 90/150/250/500 mg/kg/day) to juvenile rats from postnatal day (PND) 7 through PND 91 resulted in deaths associated with sedation and decreased male reproductive organ weights at the mid and high doses, and delayed female sexual maturation and decreased brain weights at all doses. There were no adverse effects on neurobehavioral (locomotor activity, auditory startle response, learning and memory) or reproductive function. A no-effect dose was not established. The lowest dose producing developmental toxicity in juvenile rats (20 mg/kg/day) was associated with plasma drug exposures (AUC) less than that in pediatric patients at the maximum recommended human dose (MRHD) of 1800 mg.
Oral administration of ganaxolone (0, 10, 45, or 90 mg/kg/day) to rats on postnatal day (PND) 7 resulted in widespread neuronal death in multiple brain regions, including cortex, thalamus, and hippocampus, at all doses. The pattern and extent of neuronal death was similar to that produced by intraperitoneal injection of the positive control, the NMDA receptor antagonist MK-801 (1 mg/kg). The hippocampus, in particular, is recognized to have an important role in learning and memory. Effects of ganaxolone and MK-801 on neurobehavioral function were not assessed in this study. Brain development on PND 7 in rat corresponds to that beginning in humans during the third trimester of pregnancy and continuing for the first several months to years after birth. At the lowest-effect dose for neuronal death, plasma drug exposure in the neonatal rat was less than that in pediatric patients at the MRHD.
Oral administration of M2 (0, 15, 30, or 60 mg/kg/day), a human-specific metabolite of ganaxolone, to juvenile rats from PNDs 7 to 70 resulted in adverse effects on reproductive system development in both sexes at all doses. Effects in females included decreased corpora lutea and follicular cysts in the ovary at the high dose and signs of precocious puberty and estrous cycle disruption at all doses. Female reproductive tract changes indicative of estrous cycle disruption persisted after a 5-week recovery period at the high dose. In males, effects consisted of delayed sexual maturation and decreased epididymis and prostate weights at the high dose, seminal vesicle atrophy at mid and high doses, and decreased seminal vesicle weights at all doses. Plasma exposures (AUC) of M2 at the highest dose were 2 times those in humans at the ganaxalone MRHD of 1,800 mg, while exposures at the mid and low doses were below those in humans at the ganaxolone MRHD.
Geriatric Use
CDD is largely a disease of pediatric and young adult patients. Clinical studies of ZTALMY did not include patients 65 years of age and older.
Hepatic Impairment
Administration of ZTALMY in patients with severe hepatic impairment (Child-Pugh class C) results in elevated ganaxolone plasma concentrations [see Clinical Pharmacology (12.3) ]. Therefore, dosage adjustment in these patients during titration and maintenance is required [see Dosage and Administration (2.3) ] .
No dosage adjustment is necessary in patients with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Somnolence and Sedation: Monitor for somnolence and sedation and advise patients not to drive or operate machinery until they have gained sufficient experience with ZTALMY. Concomitant use with other CNS depressants or alcohol could potentiate adverse effects. (5.1 )
- Suicidal Behavior and Ideation: Monitor patients for suicidal behavior and thoughts. (5.2 )
- Withdrawal of Antiepileptic Drugs: ZTALMY should be withdrawn gradually to minimize the risk of increased seizure frequency and status epilepticus. (5.3 )
Somnolence and Sedation
ZTALMY can cause somnolence and sedation. In Study 1 [see Clinical Studies (14) ] , the incidence of somnolence and sedation was 44% in patients treated with ZTALMY, compared with 24% in patients receiving placebo. Somnolence and sedation appeared early during treatment and were generally dose-related [see Adverse Reactions (6.1) ] .
Other central nervous system (CNS) depressants, including opioids, antidepressants, and alcohol, could potentiate somnolence and sedation in patients receiving ZTALMY [see Clinical Pharmacology (12.3) ] . Prescribers should monitor patients for somnolence and sedation, and advise patients not to drive or operate machinery until they have gained sufficient experience on ZTALMY to gauge whether it adversely affects their ability to drive or operate machinery.
Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including ZTALMY, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with an AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs, that did not include ZTALMY, showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were 4 suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5–100 years) in the clinical trials analyzed. Table 5 shows absolute and relative risk by indication for all evaluated AEDs.
| Indication | Placebo Patients with Events per 1000 Patients | Drug Patients with Events per 1000 Patients | Relative Risk: Incidence of Events in Drug Patients/ Incidence in Placebo Patients | Risk Difference: Additional Drug Patients with Events per 1000 Patients |
|---|---|---|---|---|
| Epilepsy | 1.0 | 3.4 | 3.5 | 2.4 |
| Psychiatric | 5.7 | 8.5 | 1.5 | 2.9 |
| Other | 1.0 | 1.8 | 1.9 | 0.9 |
| Total | 2.4 | 4.3 | 1.8 | 1.9 |
The relative risk for suicidal thoughts or behavior was higher in clinical trials in patients with epilepsy than in clinical trials in patients with psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing ZTALMY, or any other AED must balance the risk of suicidal thoughts or behaviors with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Withdrawal of Antiepileptic Drugs
As with most AEDs, ZTALMY should be withdrawn gradually because of the risk of increased seizure frequency and status epilepticus [see Dosage and Administration (2.4) ] . If withdrawal is needed because of a serious adverse event, rapid discontinuation can be considered.
ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in the labeling:
- Somnolence and Sedation [see Warnings and Precautions (5.1) ]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.2) ]
- Withdrawal of Antiepileptic Drugs [see Warnings and Precautions (5.3) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In controlled and uncontrolled trials in patients with seizures associated with CDD, 102 patients were treated with ZTALMY, including 83 patients treated for more than 6 months, and 50 patients treated for more than 1 year.
In Study 1, 50 patients received ZTALMY [see Clinical Studies (14) ] . The duration of treatment in this trial was up to 17 weeks. Approximately 78% of these patients were female, 92% were White, and the mean age was 6.8 years (range 2 to 19 years). All patients receiving ZTALMY, except 1, were taking other AEDs. Adverse reactions in these patients are presented below.
The most common adverse reactions (an incidence of at least 5% and at least twice the rate of placebo) were somnolence, pyrexia, salivary hypersecretion, and seasonal allergy (Table 6).
The adverse reactions leading to treatment discontinuation in ZTALMY-treated patients were somnolence and seizure (1 patient) and seizure (1 patient).
Twenty-two percent of ZTALMY-treated patients had dosing interrupted or reduced because of any adverse reaction, compared to 16% of placebo-treated patients. The most frequent adverse reactions leading to a dose interruption or reduction in ZTALMY-treated patients were somnolence (10%) and sedation (2%).
Table 6 presents the adverse reactions that occurred in ZTALMY-treated patients with seizures associated with CDD at a rate of at least 3% and at a rate greater than in placebo-treated patients during the double-blind phase.
| Adverse Reactions | ZTALMY (N=50) | Placebo (N=51) |
|---|---|---|
| % | % | |
| Somnolence somnolence includes the terms lethargy and hypersomnia adverse reactions that occurred with a 21-day titration; somnolence and sedation are expected to be reduced with the recommended 28-day titration [see Dosage and Administration (2.1, 2.3)] | 38 | 20 |
| Pyrexia | 18 | 8 |
| Upper respiratory tract infection | 10 | 6 |
| Sedation | 6 | 4 |
| Salivary Hypersecretion | 6 | 2 |
| Seasonal allergy | 6 | 0 |
| Bronchitis | 4 | 0 |
| Influenza | 4 | 2 |
| Gait disturbance | 4 | 2 |
| Nasal congestion | 4 | 2 |
DRUG INTERACTIONS
- UGT inhibitors may increase ganaxolone exposures; consider reduction of a stable ZTALMY dosage when initiating a UGT inhibitor. (7.1 )
- Cytochrome P450 inducers will decrease ganaxolone exposure. It is recommended to avoid concomitant use with strong or moderate CYP3A4 inducers; if unavoidable, consider a dosage increase of ZTALMY, but do not exceed the maximum recommended dosage. (7.2 )
Effect of UGT Inhibitors on ZTALMY
Concomitant use of ZTALMY and UGT inhibitors (e.g., valproic acid) may increase the exposure of ganaxolone, which may increase the risk of ZTALMY associated adverse reactions in patients who have titrated to a stable ZTALMY dosage. Consider a reduction of ZTALMY maintenance dosage when initiating a UGT inhibitor [see Clinical Pharmacology (12.3) ].
Effect of Strong or Moderate Cytochrome P450 Inducers on ZTALMY
Coadministration of ZTALMY with CYP450 inducers, such as strong or moderate CYP3A4 inducers, will decrease ganaxolone exposure, which can lower the efficacy of ZTALMY [see Clinical Pharmacology (12.3) ] .
It is recommended to avoid concomitant use of strong or moderate CYP3A4 inducers with ZTALMY. When concomitant use of strong or moderate CYP3A4 inducers is unavoidable, consider an increase in the dosage of ZTALMY; however, do not exceed the maximum daily dosage of ZTALMY [see Dosage and Administration (2.1) ] .
In patients on a stable ZTALMY dosage who are initiating or increasing the dosages of enzyme-inducing antiepileptic drugs (e.g., carbamazepine, phenytoin, phenobarbital, and primidone), the ZTALMY dosage may need to be increased; however, do not exceed the maximum daily dosage of ZTALMY [see Dosage and Administration (2.1) ] .
Concomitant Use of ZTALMY with CNS Depressants and Alcohol
Concomitant use of ZTALMY with CNS depressants, including alcohol, may increase the risk of somnolence and sedation [see Warnings and Precautions (5.1) ] .
DESCRIPTION
ZTALMY (ganaxolone) oral suspension contains ganaxolone, a neuroactive steroid gamma-aminobutyric acid A (GABA A ) receptor positive modulator. Ganaxolone (1-[(3R, 5S, 8R, 9S, 10S, 13S, 14S, 17S)-3-hydroxy-3, 10, 13-trimethyl-1, 2, 4, 5, 6, 7, 8, 9, 11, 12, 14, 15, 16, 17-tetradecahydrocyclopenta[a]phenanthren-17-yl]ethanone) is a methyl-substituted (at the 3β position) analog of the endogenous neurosteroid allopregnanolone, a derivative of progesterone. Its empirical formula is C 22 H 36 O 2 , and the molecular weight is 332.53 g/mol. The chemical structure is:

Ganaxolone is a white to off-white crystalline powder that only exists in one crystal form and has low aqueous solubility.
ZTALMY is an oral suspension of ganaxolone. Each mL of oral suspension contains 50 mg of ganaxolone. Inactive ingredients include artificial cherry flavor, citric acid, hypromellose, methylparaben, polyvinyl alcohol, propylparaben, purified water, simethicone emulsion, sodium benzoate, sodium citrate, sodium lauryl sulfate, and sucralose.
CLINICAL PHARMACOLOGY
Mechanism of Action
The precise mechanism by which ganaxolone exerts its therapeutic effects in the treatment of seizures associated with CDD is unknown, but its anticonvulsant effects are thought to result from positive allosteric modulation of the gamma-aminobutyric acid type A (GABA A ) receptor in the CNS.
Pharmacodynamics
Cardiac Electrophysiology
At therapeutic exposures, ZTALMY does not prolong the QTc interval. QT effects of ZTALMY at high clinical exposure scenarios have not been evaluated.
Pharmacokinetics
Absorption
Following oral administration of ZTALMY, ganaxolone is absorbed with a time to maximum plasma concentration (T max ) of 2 to 3 hours.
Effect of Food
When ZTALMY was administered with a high-fat meal, the C max and AUC increased by 3- and 2-fold, respectively, when compared to administration under fasted conditions. ZTALMY was administered with food in the clinical efficacy study, Study 1 [see Dosage and Administration (2.1) ] . The efficacy of ZTALMY when administered in the fasted state is unknown.
Distribution
Ganaxolone is approximately 99% protein-bound in serum.
Elimination
The terminal half-life for ganaxolone is 34 hours.
Metabolism
Ganaxolone is metabolized by CYP3A4/5, CYP2B6, CYP2C19, CYP2D6, UGT1A3, UGT1A6, UGT1A9, UGT2B7, and UGT2B15.
Excretion
Following a single oral dose of 300 mg [ 14 C]-ganaxolone to healthy male subjects, 55% of the total radioactivity was recovered in feces (2% as unchanged ganaxolone) and 18% of the total radioactivity dose was recovered in urine (undetected as unchanged ganaxolone).
Specific Populations
Age, sex, and race are not expected to have a clinically-relevant effect on ganaxolone pharmacokinetics, after accounting for body weight.
Pediatric Patients
After accounting for body weight, the observed pharmacokinetic exposures in patients in Study 1 [see Clinical Studies (14) ] were comparable across the age groups 2 to less than 6 years of age (n=45), 6 to less than 12 years of age (n=28), and 12 to less than 18 years of age (n=16).
Patients with Renal Impairment
Following oral administration of a single 300 mg dose of ZTALMY in subjects with severe renal impairment (creatine clearance between 15 and 30 mL/min as estimated by Cockcroft-Gault formula), the AUC 0-INF of ganaxolone decreased 8% and C max decreased 11% as compared to that in subjects with normal renal function (creatinine clearance ≥ 90 mL/min as estimated by Cockcroft-Gault formula). The changes in ganaxolone exposures when administered in patients with impaired renal function (creatinine clearance <90 mL/min) are not expected to be clinically significant.
Patients with Hepatic Impairment
The influence of hepatic impairment on the pharmacokinetics of ganaxolone was studied following a single oral dose of ZTALMY 300 mg. In subjects with mild hepatic impairment (Child-Pugh class A), C max and AUC last increased by 38% and 8%, respectively, compared to subjects with normal hepatic function. In subjects with moderate hepatic impairment (Child-Pugh class B), C max and AUC last increased by 45% and 50%, respectively, compared to subjects with normal hepatic function. In subjects with severe hepatic impairment (Child-Pugh class C), C max and AUC last increased by 148% and 269%, respectively, compared to subjects with normal hepatic function [see Dosage and Administration (2.3) and Use in Specific Populations (8.6) ] .
Drug Interaction Studies
Clinical Studies
CYP3A4 Inducers
Coadministration of ZTALMY with rifampin, a strong inducer of CYP2C19 and CYP3A4, and a moderate inducer of CYP2B6, decreased C max and AUC of ganaxolone by 57% and 68%, respectively, in healthy subjects [see Drug Interactions (7.2) ] . No dedicated drug-interaction studies were conducted with moderate or weak CYP3A4 inducers.
CYP3A4 Inhibitors
Coadministration of ZTALMY with itraconazole, a strong CYP3A4 inhibitor, increased the AUC of ganaxolone by 17% in healthy subjects (C max was unchanged). Changes in ganaxolone exposures when coadministered with strong, moderate, or weak CYP3A4 inhibitors are not expected to be clinically significant.
CYP3A4 Substrates
Coadministration of ganaxolone at steady state (400 mg twice daily; 0.44 times the maximum recommended dosage) with midazolam, a sensitive CYP3A4 substrate, did not result in clinically relevant changes in exposures of the substrate in healthy subjects.
In Vitro Studies
CYP450 Enzymes
Ganaxolone does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5 at clinically relevant concentrations. Ganaxolone does not induce CYP1A2, CYP2B6, or CYP3A4 at clinically relevant concentrations.
UGT Enzymes
Ganaxolone is a substrate for UGT1A3, UGT1A6, UGT1A9, UGT2B7, and UGT2B15. Ganaxolone does not inhibit UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, or UGT2B7.
Transporter Systems
Ganaxolone is not a substrate of BCRP, P-gp, OCT1, OCT2, OATP1B1, or OATP1B3 at clinically relevant concentrations. Ganaxolone does not inhibit BCRP, P-gp, MATE1, MATE2-K, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, or BSEP at clinically relevant concentrations.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral administration of ganaxolone (150, 500, and 1000 mg/kg/day in males and 100, 300, and 1000 mg/kg/day in females) to Tg.rasH2 mice for up to 26 weeks did not result in an increase in tumors.
No carcinogenicity studies have been conducted with ganaxolone in rats.
Mutagenesis
Ganaxolone was negative for genotoxicity in in vitro (Ames and mouse lymphoma) and in vivo (rat bone marrow micronucleus) assays. The major circulating human metabolite, oxy-dehydro-ganaxolone, was negative for mutagenicity in the in vitro Ames assay but positive for clastogenicity in an in vitro mammalian chromosomal aberration test in human peripheral blood lymphocytes.
Impairment of Fertility
Oral administration of ganaxolone (0, 10, 20 or 40 mg/kg/day) to male and female rats prior to and throughout mating and continuing in females during early gestation resulted in alterations in estrous cyclicity at the high dose. There were no effects on spermatogenesis, reproductive performance and fertility, or early embryonic development. The highest dose tested (40 mg/kg/day) was associated with plasma exposures (AUC) less than that in adult humans at the maximum recommended human dose of 1800 mg.
CLINICAL STUDIES
The effectiveness of ZTALMY for the treatment of seizures associated with CDD in patients 2 years of age and older was established in a single, double-blind, randomized, placebo-controlled study in patients 2 to 19 years of age (Study 1, NCT03572933).
Patients enrolled in Study 1 (N=50 for ZTALMY; N=51 for placebo) had molecular confirmation of a pathogenic or likely pathogenic mutation in the CDKL5 gene, seizures inadequately controlled by at least 2 previous treatment regimens, and a minimum of 16 major motor seizures (i.e., bilateral tonic, generalized tonic-clonic, bilateral clonic, atonic, focal to bilateral tonic-clonic) per 28 days during a retrospective 2-month period prior to screening.
Patients were randomized in a 1:1 ratio to receive either ZTALMY or placebo. Following a titration period, patients in the ZTALMY arm weighing 28 kg or less received a maintenance dosage of 21 mg/kg three times daily (with a maximum daily dose of 1800 mg) while patients in the ZTALMY arm weighing more than 28 kg received a maintenance dosage of 600 mg three times daily [see Dosage and Administration (2.1) ].
Ninety-six percent of patients were taking between 1 to 4 concomitant AEDs. The most frequently used concomitant AEDs (in at least 20% of patients) were valproate (34%), levetiracetam (26%), clobazam (25%), and vigabatrin (22%).
The primary efficacy endpoint was the percentage change in the 28-day frequency of major motor seizures (defined similarly as in the 2-month period prior to screening) from a 6-week prospective baseline phase during the 17-week double-blind phase. Patients treated with ZTALMY had a significantly greater reduction in the 28-day frequency of major motor seizures compared to patients receiving placebo (see Table 7 ).
| Frequency of Major Motor Seizures (per 28 days) | Placebo (N=51) | ZTALMY (N=49) |
|---|---|---|
| Prospective Baseline Phase Median Seizure Frequency | 49 | 54 |
| Median Percent Change from Baseline During Treatment | -7 | -31 |
| p-value compared to placebo Obtained from a Wilcoxon rank-sum test | 0.0036 |
Figure 1 displays the percentage of patients by category of reduction from baseline in the 28-day frequency of major motor seizures during the 17-week double-blind phase.
Figure 1 Proportion of Patients by Category of Seizure Response for ZTALMY and Placebo in Patients with CDD (Study 1)

HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ZTALMY (ganaxolone) oral suspension (50 mg/mL) is a cherry flavored white to off-white suspension supplied in a 4 fl. oz (135 mL) round natural high density polyethylene (HDPE) bottle with a propylene child-resistant cap containing 110 mL of ZTALMY oral suspension.
ZTALMY is packaged in a carton with 1 bottle (NDC 81583-100-01).
Storage and Handling
Store ZTALMY in its original bottle in an upright position at 20°C to 25°C (68°F to 77°F); excursions permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Keep the cap tightly closed. Use within 30 days of first opening the bottle, then discard any remainder.
INSTRUCTIONS FOR USE
ZTALMY ® ( zuh-tal' mee ) (ganaxolone) oral suspension, CV 50 mg/mL
Be sure that you read, understand, and follow these instructions carefully to ensure proper dosing of the oral suspension.
Important:
- Follow your healthcare provider's instructions for how to take or give ZTALMY.
- ZTALMY should always be given with food.
- Ask your healthcare provider or pharmacist if you are not sure how to prepare, take, or give the prescribed dose of ZTALMY.
- Always use the oral syringe provided by your pharmacist to make sure you measure the right amount of ZTALMY.
- Do not use ZTALMY after the expiration date on the package and each bottle.
- Use ZTALMY within 30 days of first opening the bottle.
- After 30 days of first opening the bottle, safely throw away (dispose of) any ZTALMY that has not been used.
Each package contains:
| 1 bottle of ZTALMY with a child resistant cap: |
|

Supplies not included in the package:
- press-in bottle adapter
- oral syringe
You can get a press-in bottle adapter and oral syringes from your pharmacy. Your pharmacist can help you choose the right press-in bottle adapter and oral syringe to use with ZTALMY.
Call your pharmacist right away if you do not have a press-in bottle adapter and the right sized oral syringe to use with your medicine.
Note: If you lose or damage an oral syringe, or cannot read the markings, contact your pharmacist for a new oral syringe.
Follow the instructions below to use the press-in bottle adapter and oral syringe to measure and take or give ZTALMY.
Prepare the bottle
|
|
|
|
|
|
|
|




Prepare the dose
Your healthcare provider will tell you how much ZTALMY to take or give.
|
|
|
|
|
|
|
|





Take or give ZTALMY
|
|
|
|

How should I store ZTALMY?
- Store ZTALMY at 68°F to 77°F (20°C to 25°C).
- Always store ZTALMY in its original bottle in an upright position.
- Keep the child-resistant cap tightly closed.
- Use within 30 days of first opening the bottle. Throw away (dispose of) any unused medicine after 30 days.
Keep ZTALMY and all medicines out of the reach of children.
Helpline Details
For additional assistance, call the toll-free helpline at 844-IMMEDICA (1-844-627-4687). Hours: Monday - Friday 8:00 am to 6:00 pm EST
Frequently asked Questions:
| Q: | What if there are air bubbles in the oral syringe? |
| A: | Slowly push the liquid back into the bottle and repeat step 6 until the air bubbles are gone. |
Manufactured for Immedica Pharma AB, Stockholm, Sweden Marketed by Immedica Pharma US Inc., Chicago, IL 60642 For more information, go to www.ZTALMY.com or call 1-484-801-4670. © Immedica Pharma US Inc. All rights reserved. ZTALMY is a registered trademark of Immedica Pharma US Inc.
Issued: 11/2025
Mechanism of Action
The precise mechanism by which ganaxolone exerts its therapeutic effects in the treatment of seizures associated with CDD is unknown, but its anticonvulsant effects are thought to result from positive allosteric modulation of the gamma-aminobutyric acid type A (GABA A ) receptor in the CNS.