Get your patient on Diclofenac Sodium - Diclofenac Sodium tablet, Delayed Release (Diclofenac Sodium)
Diclofenac Sodium - Diclofenac Sodium tablet, Delayed Release prescribing information
WARNING: RISK OF SERIOUS CARDIOVASCULAR AND GASTROINTESTINAL EVENTS
Cardiovascular Thrombotic Events
- Nonsteroidal anti-inflammatory drugs (NSAIDs) cause an increased risk of serious cardiovascular thrombotic events, including myocardial infarction and stroke, which can be fatal. This risk may occur early in treatment and may increase with duration of use (see WARNINGS ).
- Diclofenac sodium delayed-release tablets are contraindicated in the setting of coronary artery bypass graft (CABG) surgery (see CONTRAINDICATIONS and WARNINGS ).
Gastrointestinal Bleeding, Ulceration, and Perforation
- NSAIDs cause an increased risk of serious gastrointestinal (GI) adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly patients and patients with a prior history of peptic ulcer disease and/or GI bleeding are at greater risk for serious GI events (see WARNINGS ).
INDICATIONS AND USAGE
Carefully consider the potential benefits and risks of diclofenac sodium delayed-release tablets and other treatment options before deciding to use diclofenac sodium delayed-release tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS: Gastrointestinal Bleeding, Ulceration and Perforation ).
Diclofenac sodium delayed-release tablets are indicated:
- For relief of the signs and symptoms of osteoarthritis
- For relief of the signs and symptoms of rheumatoid arthritis
- For acute or long-term use in the relief of signs and symptoms of ankylosing spondylitis
DOSAGE AND ADMINISTRATION
Carefully consider the potential benefits and risks of diclofenac sodium delayed-release tablets and other treatment options before deciding to use diclofenac sodium delayed-release tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS: Gastrointestinal Bleeding, Ulceration, and Perforation ).
After observing the response to initial therapy with diclofenac sodium delayed-release tablets, the dose and frequency should be adjusted to suit an individual patient’s needs.
For the relief of osteoarthritis, the recommended dosage is 100 to 150 mg/day in divided doses (50 mg twice a day or three times a day, or 75 mg twice a day).
For the relief of rheumatoid arthritis, the recommended dosage is 150 to 200 mg/day in divided doses (50 mg three times a day or four times a day, or 75 mg twice a day).
For the relief of ankylosing spondylitis, the recommended dosage is 100 to 125 mg/day, administered as 25 mg four times a day, with an extra 25 mg dose at bedtime if necessary.
Different formulations of diclofenac (diclofenac sodium enteric-coated tablets; diclofenac sodium extended-release tablets; diclofenac potassium immediate-release tablets) are not necessarily bioequivalent even if the milligram strength is the same.
CONTRAINDICATIONS
Diclofenac sodium delayed-release tablets are contraindicated in the following patients.
- Known hypersensitivity (e.g., anaphylactic reactions and serious skin reactions) to diclofenac or any components of the drug product (see WARNINGS: Anaphylactic Reactions , Serious Skin Reactions ).
- History of asthma, urticaria, or other allergic-type reactions after taking aspirin or other NSAIDs. Severe, sometimes fatal, anaphylactic reactions to NSAIDs have been reported in such patients (see WARNINGS: Anaphylactic Reaction , Exacerbation of Asthma Related to Aspirin Sensitivity ).
- In the setting of coronary artery bypass graft (CABG) surgery (see WARNINGS: Cardiovascular Thrombotic Events ).
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Cardiovascular Thrombotic Events (see WARNINGS )
- GI Bleeding, Ulceration and Perforation (see WARNINGS )
- Hepatotoxicity (see WARNINGS )
- Hypertension (see WARNINGS )
- Heart Failure and Edema (see WARNINGS )
- Renal Toxicity and Hyperkalemia (see WARNINGS )
- Anaphylactic Reactions (see WARNINGS )
- Serious Skin Reactions (see WARNINGS )
- Hematologic Toxicity (see WARNINGS )
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In patients taking diclofenac sodium delayed-release tablets, or other NSAIDs, the most frequently reported adverse experiences occurring in approximately 1% to 10% of patients are:
Gastrointestinal experiences including: abdominal pain, constipation, diarrhea, dyspepsia, flatulence, gross bleeding/perforation, heartburn, nausea, GI ulcers (gastric/duodenal) and vomiting.
Abnormal renal function, anemia, dizziness, edema, elevated liver enzymes, headaches, increased bleeding time, pruritus, reactions and tinnitus.
Additional adverse experiences reported occasionally include:
Body as a Whole fever, infection, sepsis
Cardiovascular System congestive heart failure, hypertension, tachycardia, syncope
Digestive System dry mouth, esophagitis, gastric/peptic ulcers, gastritis, gastrointestinal bleeding, glossitis, hematemesis, hepatitis, jaundice
Hemic and Lymphatic System
ecchymosis, eosinophilia, leukopenia, melena, purpura, rectal bleeding, stomatitis, thrombocytopenia
Metabolic and Nutritional weight changes
Nervous System
anxiety, asthenia, confusion, depression, dream abnormalities, drowsiness, insomnia, malaise, nervousness, paresthesia, somnolence, tremors, vertigo
Respiratory System
asthma, dyspnea
Skin and Appendages
alopecia, photosensitivity, sweating increased
Special Senses
blurred vision
Urogenital System
cystitis, dysuria, hematuria, interstitial nephritis, oliguria/polyuria, proteinuria, renal failure
Other adverse reactions, which occur rarely are:
Body as a Whole
anaphylactic reactions, appetite changes, death
Cardiovascular System
arrhythmia, hypotension, myocardial infarction, palpitations, vasculitis
Digestive System
colitis, eructation, fulminant hepatitis with and without jaundice, liver failure, liver necrosis, pancreatitis
Hemic and Lymphatic System
agranulocytosis, hemolytic anemia, aplastic anemia, lymphadenopathy, pancytopenia
Metabolic and Nutritional
hyperglycemia
Nervous System
convulsions, coma, hallucinations, meningitis
Respiratory System
respiratory depression, pneumonia
Skin and Appendages
angioedema, toxic epidermal necrolysis, erythema multiforme, exfoliative dermatitis, Stevens-Johnson syndrome, fixed drug eruption (FDE), urticaria
Special Senses
conjunctivitis, hearing impairment
To report SUSPECTED ADVERSE REACTIONS , contact Rising Pharma Holdings, Inc. at 1-844-874-7464 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
See Table 2 for clinically significant drug interactions with diclofenac.
| Drugs That Interfere with Hemostasis | |
| Clinical Impact: |
|
| Intervention: | Monitor patients with concomitant use of diclofenac sodium delayed-release tablets with anticoagulants (e.g., warfarin), antiplatelet agents (e.g., aspirin), selective serotonin reuptake inhibitors (SSRIs), and serotonin norepinephrine reuptake inhibitors (SNRIs) for signs of bleeding (see WARNINGS: Hematologic Toxicity ). |
| Aspirin | |
| Clinical Impact: | Controlled clinical studies showed that the concomitant use of NSAIDs and analgesic doses of aspirin does not produce any greater therapeutic effect than the use of NSAIDs alone. In a clinical study, the concomitant use of an NSAID and aspirin was associated with a significantly increased incidence of GI adverse reactions as compared to use of the NSAID alone (see WARNINGS: Gastrointestinal Bleeding, Ulceration, and Perforation ). |
| Intervention: | Concomitant use of diclofenac sodium delayed-release tablets and analgesic doses of aspirin is not generally recommended because of the increased risk of bleeding (see WARNINGS: Hematologic Toxicity ). Diclofenac sodium delayed-release tablets are not a substitute for low dose aspirin for cardiovascular protection. |
| ACE Inhibitors, Angiotensin Receptor Blockers, and Beta-Blockers | |
| Clinical Impact: |
|
| Intervention: |
|
| Diuretics | |
| Clinical Impact: | Clinical studies, as well as post-marketing observations, showed that NSAIDs reduced the natriuretic effect of loop diuretics (e.g., furosemide) and thiazide diuretics in some patients. This effect has been attributed to the NSAID inhibition of renal prostaglandin synthesis. |
| Intervention: | During concomitant use of diclofenac sodium delayed-release tablets with diuretics, observe patients for signs of worsening renal function, in addition to assuring diuretic efficacy including antihypertensive effects (see WARNINGS: Renal Toxicity and Hyperkalemia ). |
| Digoxin | |
| Clinical Impact: | The concomitant use of diclofenac with digoxin has been reported to increase the serum concentration and prolong the half-life of digoxin. |
| Intervention: | During concomitant use of diclofenac sodium delayed-release tablets and digoxin, monitor serum digoxin levels. |
| Lithium | |
| Clinical Impact: | NSAIDs have produced elevations in plasma lithium levels and reductions in renal lithium clearance. The mean minimum lithium concentration increased 15%, and the renal clearance decreased by approximately 20%. This effect has been attributed to NSAID inhibition of renal prostaglandin synthesis. |
| Intervention: | During concomitant use of diclofenac sodium delayed-release tablets and lithium, monitor patients for signs of lithium toxicity. |
| Methotrexate | |
| Clinical Impact: | Concomitant use of NSAIDs and methotrexate may increase the risk for methotrexate toxicity (e.g., neutropenia, thrombocytopenia, renal dysfunction). |
| Intervention: | During concomitant use of diclofenac sodium delayed-release tablets and methotrexate, monitor patients for methotrexate toxicity. |
| Cyclosporine | |
| Clinical Impact: | Concomitant use of diclofenac sodium delayed-release tablets and cyclosporine may increase cyclosporine’s nephrotoxicity. |
| Intervention: | During concomitant use of diclofenac sodium delayed-release tablets and cyclosporine, monitor patients for signs of worsening renal function. |
| NSAIDs and Salicylates | |
| Clinical Impact: | Concomitant use of diclofenac with other NSAIDs or salicylates (e.g., diflunisal, salsalate) increases the risk of GI toxicity, with little or no increase in efficacy (see WARNINGS: Gastrointestinal Bleeding, Ulceration, and Perforation ). |
| Intervention: | The concomitant use of diclofenac with other NSAIDs or salicylates is not recommended. |
| Pemetrexed | |
| Clinical Impact: | Concomitant use of diclofenac sodium delayed-release tablets and pemetrexed may increase the risk of pemetrexed- associated myelosuppression, renal, and GI toxicity (see the pemetrexed prescribing information). |
| Intervention: | During concomitant use of diclofenac sodium delayed-release tablets and pemetrexed, in patients with renal impairment whose creatinine clearance ranges from 45 to 79 mL/min, monitor for myelosuppression, renal and GI toxicity. NSAIDs with short elimination half-lives (e.g., diclofenac, indomethacin) should be avoided for a period of two days before, the day of, and two days following administration of pemetrexed. In the absence of data regarding potential interaction between pemetrexed and NSAIDs with longer half-lives (e.g., meloxicam, nabumetone), patients taking these NSAIDs should interrupt dosing for at least five days before, the day of, and two days following pemetrexed administration. |
| CYP2C9 Inhibitors or Inducers: | |
| Clinical Impact: | Diclofenac is metabolized by cytochrome P450 enzymes, predominantly by CYP2C9. Co-administration of diclofenac with CYP2C9 inhibitors (e.g. voriconazole) may enhance the exposure and toxicity of diclofenac whereas co-administration with CYP2C9 inducers (e.g. rifampin) may lead to compromised efficacy of diclofenac. |
| Intervention: | A dosage adjustment may be warranted when diclofenac is administered with CYP2C9 inhibitors or inducers (see CLINICAL PHARMACOLOGY: Pharmacokinetics ). |
DESCRIPTION
Diclofenac sodium is a benzeneacetic acid derivative, designated chemically as 2-[(2,6-dichlorophenyl)amino] benzeneacetic acid, monosodium salt. The structural formula is:
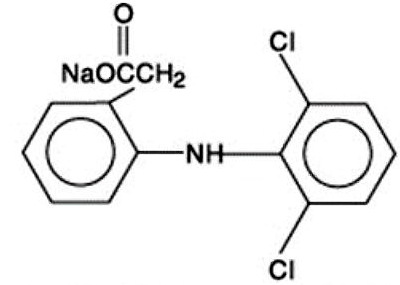
C 14 H 10 Cl 2 NNaO 2 M.W. 318.14
Diclofenac sodium is a white to off-white, hygroscopic crystalline powder. It is freely soluble in methanol, soluble in ethanol, sparingly soluble in water and practically insoluble in chloroform and in dilute acid. The n-octanol/water partition coefficient is 13.4 at pH 7.4 and 1545 at pH 5.2. Diclofenac sodium has a dissociation constant (pKa) of 4.0 ± 0.2 at 25°C in water.
Each enteric-coated tablet for oral administration contains 25 mg, 50 mg, or 75 mg of diclofenac sodium. In addition, each tablet contains the following inactive ingredients. Inactive ingredients: lactose (monohydrate), microcrystalline cellulose, croscarmellose sodium, povidone, talc, magnesium stearate, methacrylic acid copolymer, polyethylene glycol, titanium dioxide, hypromellose, iron oxide red, iron oxide yellow.
CLINICAL PHARMACOLOGY
Mechanism of Action
Diclofenac has analgesic, anti-inflammatory, and antipyretic properties.
The mechanism of action of diclofenac sodium delayed-release tablets, like that of other NSAIDs, is not completely understood but involves inhibition of cyclooxygenase (COX-1 and COX-2).
Diclofenac is a potent inhibitor of prostaglandin synthesis in vitro . Diclofenac concentrations reached during therapy have produced in vivo effects. Prostaglandins sensitize afferent nerves and potentiate the action of bradykinin in inducing pain in animal models. Prostaglandins are mediators of inflammation. Because diclofenac is an inhibitor of prostaglandin synthesis, its mode of action may be due to a decrease of prostaglandins in peripheral tissues.
Pharmacokinetics
Absorption
Diclofenac is 100% absorbed after oral administration compared to IV administration as measured by urine recovery. However, due to first-pass metabolism, only about 50% of the absorbed dose is systemically available (see Table 1). Food has no significant effect on the extent of diclofenac absorption. However, there is usually a delay in the onset of absorption of 1 to 4.5 hours and a reduction in peak plasma levels of <20%.
| Normal Healthy Adults (20-48 years) | ||
| PK Parameter | Mean | Coefficient of Variation (%) |
| Absolute Bioavailability (%) [N = 7] | 55 | 40 |
| Tmax (hr) [N = 56] | 2.3 | 69 |
| Oral Clearance (CL/F; mL/min) [N = 56] | 582 | 23 |
| Renal Clearance (% unchanged drug in urine)[N = 7] | <1 | – |
| Apparent Volume of Distribution (V/F; L/kg) [N = 56] | 1.4 | 58 |
| Terminal Half-life (hr) [N = 56] | 2.3 | 48 |
Distribution
The apparent volume of distribution (V/F) of diclofenac sodium is 1.4 L/kg.
Diclofenac is more than 99% bound to human serum proteins, primarily to albumin. Serum protein binding is constant over the concentration range (0.15 to 105 mcg/mL) achieved with recommended doses.
Diclofenac diffuses into and out of the synovial fluid. Diffusion into the joint occurs when plasma levels are higher than those in the synovial fluid, after which the process reverses and synovial fluid levels are higher than plasma levels. It is not known whether diffusion into the joint plays a role in the effectiveness of diclofenac.
Elimination
Metabolism
Five diclofenac metabolites have been identified in human plasma and urine. The metabolites include 4’-hydroxy-, 5-hydroxy-,3’-hydroxy-, 4’,5-dihydroxy- and 3’-hydroxy-4’-methoxy diclofenac. The major diclofenac metabolite, 4’-hydroxy-diclofenac, has very weak pharmacologic activity. The formation of 4’-hydroxy- diclofenac is primarily mediated by CYP2C9. Both diclofenac and its oxidative metabolites undergo glucuronidation or sulfation followed by biliary excretion. Acylglucuronidation mediated by UGT2B7 and oxidation mediated by CYP2C8 may also play a role in diclofenac metabolism. CYP3A4 is responsible for the formation of minor metabolites, 5-hydroxy- and 3’-hydroxy-diclofenac. In patients with renal dysfunction, peak concentrations of metabolites 4’-hydroxy- and 5-hydroxy-diclofenac were approximately 50% and 4% of the parent compound after single oral dosing compared to 27% and 1% in normal healthy subjects.
Excretion
Diclofenac is eliminated through metabolism and subsequent urinary and biliary excretion of the glucuronide and the sulfate conjugates of the metabolites. Little or no free unchanged diclofenac is excreted in the urine. Approximately 65% of the dose is excreted in the urine and approximately 35% in the bile as conjugates of unchanged diclofenac plus metabolites. Because renal elimination is not a significant pathway of elimination for unchanged diclofenac, dosing adjustment in patients with mild to moderate renal dysfunction is not necessary. The terminal half-life of unchanged diclofenac is approximately 2 hours.
Special Populations
Pediatric
The pharmacokinetics of diclofenac sodium delayed-release tablets has not been investigated in pediatric patients.
Race
Pharmacokinetic differences due to race have not been identified.
Hepatic Impairment
Hepatic metabolism accounts for almost 100% of diclofenac sodium delayed-release tablets elimination, so patients with hepatic disease may require reduced doses of diclofenac sodium delayed-release tablets compared to patients with normal hepatic function.
Renal Impairment
Diclofenac pharmacokinetics has been investigated in subjects with renal insufficiency. No differences in the pharmacokinetics of diclofenac have been detected in studies of patients with renal impairment. In patients with renal impairment (inulin clearance 60 to 90, 30 to 60, and <30 mL/min; N=6 in each group), AUC values and elimination rate were comparable to those in healthy subjects.
Drug Interactions Studies
Voriconazole
When co-administered with voriconazole (inhibitor of CYP2C9, 2C19 and 3A4 enzyme), the C max and AUC of diclofenac increased by 114% and 78%, respectively (see PRECAUTIONS: Drug Interactions ).
Aspirin
When NSAIDs were administered with aspirin, the protein binding of NSAIDs were reduced, although the clearance of free NSAID was not altered. The clinical significance of this interaction is not known. See Table 2 for clinically significant drug interactions of NSAIDs with aspirin (see PRECAUTIONS: Drug Interactions ).
HOW SUPPLIED Diclofenac sodium delayed-release tablets, USP, for oral administration, are available as:
25 mg: round, light brown, enteric-coated tablets P 25 imprinted on one side in black ink and plain on the reverse side are supplied as: Bottles of 30........................................ NDC 16571-203-03
Bottles of 100.......................................NDC 16571-203-10
50 mg: round, light brown, enteric-coated tablets P 50 imprinted on one side in black ink and plain on the reverse side are supplied as: Bottles of 60.........................................NDC 16571-202-06 Bottles of 100.......................................NDC 16571-202-10 Bottles of 1000.....................................NDC 16571-202-11
75 mg: round, light brown, enteric-coated tablets P 75 imprinted on one side in black ink and plain on the reverse side are supplied as: Bottles of 60.........................................NDC 16571-201-06 Bottles of 100.......................................NDC 16571-201-10 Bottles of 500.......................................NDC 16571-201-50 Bottles of 1000.....................................NDC 16571-201-11
Store at 20° to 25°C (68° to 77°F) (see USP Controlled Room Temperature). Protect from moisture. Dispense in a tight, light-resistant container.
Pharmacist: Dispense with Medication Guide available at: www.risingpharma.com/Medguides/diclofenac-sodium-delayed-release-tablets.pdf
Manufactured by:
Unique Pharmaceutical Laboratories (A Div. of J. B. Chemicals & Pharmaceuticals Ltd.), Mumbai 400 030, India.
Distributed by:

Rising Pharma Holdings, Inc.
East Brunswick, NJ 08816
142054 Jul. 2025
Mechanism of Action
Diclofenac has analgesic, anti-inflammatory, and antipyretic properties.
The mechanism of action of diclofenac sodium delayed-release tablets, like that of other NSAIDs, is not completely understood but involves inhibition of cyclooxygenase (COX-1 and COX-2).
Diclofenac is a potent inhibitor of prostaglandin synthesis in vitro . Diclofenac concentrations reached during therapy have produced in vivo effects. Prostaglandins sensitize afferent nerves and potentiate the action of bradykinin in inducing pain in animal models. Prostaglandins are mediators of inflammation. Because diclofenac is an inhibitor of prostaglandin synthesis, its mode of action may be due to a decrease of prostaglandins in peripheral tissues.