Get your patient on Eulexin - Flutamide capsule (Flutamide)
Eulexin - Flutamide capsule prescribing information
WARNINGS
Hepatic Injury
There have been postmarketing reports of hospitalization and rarely death due to liver failure in patients taking Eulexin™. Evidence of hepatic injury included elevated serum transaminase levels, jaundice, hepatic encephalopathy and death related to acute hepatic failure. The hepatic injury was reversible after discontinuation of therapy in some patients. Approximately half of the reported cases occurred within the initial 3 months of treatment with Eulexin™.
Serum transaminase levels should be measured prior to starting treatment with Eulexin™. Eulexin™ is not recommended in patients whose ALT values exceed twice the upper limit of normal. Serum transaminase levels should then be measured monthly for the first 4 months of therapy, and periodically thereafter. Liver function tests also should be obtained at the first signs and symptoms suggestive of liver dysfunction, e.g., nausea, vomiting, abdominal pain, fatigue, anorexia, "flu-like" symptoms, hyperbilirubinuria, jaundice or right upper quadrant tenderness. If at any time, a patient has jaundice, or their ALT rises above 2 times the upper limit of normal, Eulexin™ should be immediately discontinued with close follow-up of liver function tests until resolution.
INDICATIONS AND USAGE
Eulexin™ capsules are indicated for use in combination with LHRH-agonists for the management of locally confined Stage B 2 -C and Stage D 2 metastatic carcinoma of the prostate.
Stage B 2 -C Prostatic Carcinoma
Treatment with Eulexin™ capsules and the goserelin acetate implant should start eight weeks prior to initiating radiation therapy and continue during radiation therapy.
Stage D 2 Metastatic Carcinoma
To achieve benefit from treatment, Eulexin™ capsules should be initiated with the LHRH-agonist and continued until progression.
DOSAGE AND ADMINISTRATION
The recommended dosage is 2 capsules 3 times a day at 8 hour intervals for a total daily dose of 750 mg.
CONTRAINDICATIONS
Eulexin™ capsules are contraindicated in patients who are hypersensitive to Eulexin™ or any component of this preparation.
Eulexin™ capsules are contraindicated in patients with severe hepatic impairment (baseline hepatic enzymes should be evaluated prior to treatment).
ADVERSE REACTIONS
Stage B 2 -C Prostatic Carcinoma
Treatment with Eulexin™ capsules and the goserelin acetate implant did not add substantially to the toxicity of radiation treatment alone. The following adverse experiences were reported during a multicenter clinical trial comparing Eulexin™ + goserelin acetate implant + radiation versus radiation alone. The most frequently reported (greater than 5%) adverse experiences are listed below:
| (n=231) Goserelin Acetate Implant + Eulexin™ + Radiation | (n=235) Radiation Only | |
|---|---|---|
| % All | % All | |
| Rectum/Large Bowel | 80 | 76 |
| Bladder | 58 | 60 |
| Skin | 37 | 37 |
| (n=231) Goserelin Acetate Implant + Eulexin™ + Radiation | (n=235) Radiation Only | |
|---|---|---|
| % All | % All | |
| Diarrhea | 36 | 40 |
| Cystitis | 16 | 16 |
| Rectal Bleeding | 14 | 20 |
| Proctitis | 8 | 8 |
| Hematuria | 7 | 12 |
Additional adverse event data were collected for the combination therapy with radiation group over both the hormonal treatment and hormonal treatment plus radiation phases of the study. Adverse experiences occurring in more than 5% of patients in this group, over both parts of the study, were hot flashes (46%), diarrhea (40%), nausea (9%), and skin rash (8%).
Stage D 2 Metastatic Carcinoma
The following adverse experiences were reported during a multicenter clinical trial comparing Eulexin™ + LHRH agonist versus placebo + LHRH agonist.
The most frequently reported (greater than 5%) adverse experiences during treatment with Eulexin™ capsules in combination with an LHRH agonist are listed in the table below. For comparison, adverse experiences seen with an LHRH agonist and placebo are also listed in the following table.
| (n=294) Eulexin™ + LHRH agonist | (n=28) Placebo + LHRH agonist | |
|---|---|---|
| % All | % All | |
| Hot Flashes | 61 | 57 |
| Loss of Libido | 36 | 31 |
| Impotence | 33 | 29 |
| Diarrhea | 12 | 4 |
| Nausea/Vomiting | 11 | 10 |
| Gynecomastia | 9 | 11 |
| Other | 7 | 9 |
| Other GI | 6 | 4 |
As shown in the table, for both treatment groups, the most frequently occurring adverse experiences (hot flashes, impotence, loss of libido) were those known to be associated with low serum androgen levels and known to occur with LHRH agonists alone.
The only notable difference was the higher incidence of diarrhea in the Eulexin™ + LHRH agonist group (12%), which was severe in 5% as opposed to the placebo + LHRH agonist (4%), which was severe in less than 1%.
In addition, the following adverse reactions were reported during treatment with Eulexin™ + LHRH agonist.
Cardiovascular System: hypertension in 1% of patients.
Central Nervous System : CNS (drowsiness/confusion/depression/anxiety/nervousness) reactions occurred in 1% of patients.
Gastrointestinal System : anorexia 4%, and other GI disorders occurred in 6% of patients.
Hematopoietic System : anemia occurred in 6%, leukopenia in 3%, and thrombocytopenia in 1% of patients.
Liver and Biliary System : hepatitis and jaundice in less than 1% of patients.
Skin : irritation at the injection site and rash occurred in 3% of patients.
Other: edema occurred in 4%, genitourinary and neuromuscular symptoms in 2%, and pulmonary symptoms in less than 1% of patients.
In addition, the following spontaneous adverse experiences have been reported during the marketing of Eulexin™: hemolytic anemia,macrocytic anemia,methemoglobinemia, sulfhemoglobinemia, photosensitivity reactions (including erythema, ulceration, bullous eruptions, and epidermal necrolysis) and urine discoloration. The urine was noted to change to an amber or yellow-green appearance which can be attributed to the Eulexin™ and/or its metabolites. Also reported were cholestatic jaundice, hepatic encephalopathy, and hepatic necrosis. The hepatic conditions were often reversible after discontinuing therapy; however, there have been reports of death following severe hepatic injury associated with use of Eulexin™.
Malignant breast neoplasms have occurred rarely in male patients being treated with Eulexin™ capsules.
Abnormal Laboratory Test Values
Laboratory abnormalities including elevated SGOT, SGPT, bilirubin values, SGGT, BUN, and serum creatinine have been reported.
Drug Interactions
Increases in prothrombin time have been noted in patients receiving long-term warfarin therapy after Eulexin™ was initiated. Therefore close monitoring of prothrombin time is recommended and adjustment of the anticoagulant dose may be necessary when Eulexin™ capsules are administered concomitantly with warfarin.
DESCRIPTION
Eulexin™ capsules contain flutamide, an acetanilid, nonsteroidal, orally active antiandrogen having the chemical name, ,,-trifluoro-2-methyl-4'-nitro- m -propionotoluidide.
Each capsule contains 125 mg flutamide. The compound is a buff to yellow powder with a molecular weight of 276.22 and the following structural formula:
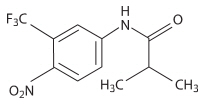
C 11 H 11 F 3 N 2 O 3
In addition, each capsule contains the following inactive ingredients: corn starch, lactose monohydrate, magnesium stearate, povidone, and sodium lauryl sulfate. Gelatin capsule shells may contain gelatin, silicon dioxide, sodium lauryl sulfate, titanium dioxide, FDA/E172 Red Iron Oxide, FDA/E172 Yellow Iron Oxide, and black ink containing pharmaceutical glaze (modified) in SD-45, synthetic black iron oxide, N-butyl alcohol, SDA-3A alcohol, FD&C Blue No.2 Aluminum Lake, FD&C Red No.40 Aluminum Lake, FD&C Blue No.1 Aluminum Lake, and D&C Yellow No.10 Aluminum Lake.
CLINICAL PHARMACOLOGY
General
In animal studies, flutamide demonstrates potent antiandrogenic effects. It exerts its antiandrogenic action by inhibiting androgen uptake and/or by inhibiting nuclear binding of androgen in target tissues or both. Prostatic carcinoma is known to be androgen-sensitive and responds to treatment that counteracts the effect of androgen and/or removes the source of androgen, e.g., castration. Elevations of plasma testosterone and estradiol levels have been noted following flutamide administration.
Pharmacokinetics
Absorption
Analysis of plasma, urine, and feces following a single oral 200 mg dose of tritium-labeled Eulexin™ to human volunteers showed that the drug is rapidly and completely absorbed. Following a single 250 mg oral dose to normal adult volunteers, the biologically active alpha-hydroxylated metabolite reaches maximum plasma concentrations in about 2 hours, indicating that it is rapidly formed from flutamide. Food has no effect on the bioavailability of flutamide.
Distribution
In male rats administered an oral 5 mg/kg dose of 14 C-flutamide neither flutamide nor any of its metabolites is preferentially accumulated in any tissue except the prostate. Total drug levels were highest 6 hours after drug administration in all tissues. Levels declined at roughly similar rates to low levels at 18 hours. The major metabolite was present at higher concentrations than Eulexin™ in all tissues studied. Following a single 250 mg oral dose to normal adult volunteers, low plasma concentrations of Eulexin™ were detected. The plasma half-life for the alpha-hydroxylated metabolite of Eulexin™ is approximately 6 hours. Eulexin™, in vivo , at steady-state plasma concentrations of 24 to 78 ng/mL, is 94% to 96% bound to plasma proteins. The active metabolite of Eulexin™, in vivo , at steady-state plasma concentrations of 1556 to 2284 ng/mL, is 92% to 94% bound to plasma proteins.
Metabolism
The composition of plasma radioactivity, following a single 200 mg oral dose of tritium-labeled Eulexin™ to normal adult volunteers, showed that Eulexin™ is rapidly and extensively metabolized, with Eulexin™ comprising only 2.5% of plasma radioactivity 1 hour after administration. At least six metabolites have been identified in plasma. The major plasma metabolite is a biologically active alpha-hydroxylated derivative which accounts for 23% of the plasma tritium 1 hour after drug administration. The major urinary metabolite is 2-amino-5nitro-4-(trifluoromethyl)phenol.
Excretion
Eulexin™ and its metabolites are excreted mainly in the urine with only 4.2% of a single dose excreted in the feces over 72 hours.
| Single Dose flutamide | Hydroxyflutamide | Steady-State flutamide | Hydroxyflutamide | |
|---|---|---|---|---|
| C max (ng/mL) | 25.2 ± 34.2 | 894 ± 406 | 113 ± 213 | 1629 ± 586 |
| Elimination half-life (hr) | — | 8.1 ± 1.3 | 7.8 | 9.6 ± 2.5 |
| T max (hr) | 1.9 ± 0.7 | 2.7 ± 1.0 | 1.3 ± 0.7 | 1.9 ± 0.6 |
| C min (ng/mL) | — | — | — | 673 ± 316 |
Special Populations
Geriatric
Following multiple oral dosing of 250 mg t.i.d. in normal geriatric volunteers, Eulexin™ and its active metabolite approached steady-state plasma levels (based on pharmacokinetic simulations) after the fourth Eulexin™ dose. The half-life of the active metabolite in geriatric volunteers after a single Eulexin™ dose is about 8 hours and at steady-state in 9.6 hours.
Race
There are no known alterations in Eulexin™ absorption, distribution, metabolism, or excretion due to race.
Renal Impairment
Following a single 250 mg dose of Eulexin™ administered to subjects with chronic renal insufficiency, there appeared to be no correlation between creatinine clearance and either C max or AUC of Eulexin™. Renal impairment did not have an effect on the C max or AUC of the biologically active alpha-hydroxylated metabolite of Eulexin™. In subjects with creatinine clearance of < 29 mL/min, the half-life of the active metabolite was slightly prolonged. Eulexin™ and its active metabolite were not well dialyzed. Dose adjustment in patients with chronic renal insufficiency is not warranted.
Hepatic Impairment
No information on the pharmacokinetics of Eulexin™ in hepatic impairment is available (see BOXED WARNINGS, Hepatic Injury ).
Women, Pediatrics
Eulexin™ has not been studied in women or pediatric subjects.
Drug-Drug Interactions
Interactions between Eulexin™ capsules and LHRH-agonists have not occurred. Increases in prothrombin time have been noted in patients receiving warfarin therapy (see PRECAUTIONS ).
CLINICAL STUDIES
Eulexin™ has been demonstrated to interfere with testosterone at the cellular level. This can complement medical castration achieved with LHRH-agonists which suppresses testicular androgen production by inhibiting luteinizing hormone secretion.
The effects of combination therapy have been evaluated in two studies. One study evaluated the effects of Eulexin™ and an LHRH-agonist as neoadjuvant therapy to radiation in stage B 2 -C prostatic carcinoma and the other study evaluated Eulexin™ and an LHRH-agonist as the sole therapy in stage D 2 prostatic carcinoma.
Stage B 2 -C Prostatic Carcinoma
The effects of hormonal treatment combined with radiation was studied in 466 patients (231 Eulexin™ capsules + goserelin acetate implant + radiation, 235 radiation alone) with bulky primary tumors confined to the prostate (stage B 2 ) or extending beyond the capsule (stage C), with or without pelvic node involvement.
In this multicentered, controlled trial, administration of Eulexin™ capsules (250 mg t.i.d.) and goserelin acetate (3.6 mg depot) prior to and during radiation was associated with a significantly lower rate of local failure compared to radiation alone (16% vs. 33% at 4 years, P < 0.001). The combination therapy also resulted in a trend toward reduction in the incidence of distant metastases (27% vs. 36% at 4 years, P = 0.058). Median disease-free survival was significantly increased in patients who received complete hormonal therapy combined with radiation as compared to those patients who received radiation alone (4.4 vs 2.6 years, P < 0.001). Inclusion of normal PSA level as a criterion for disease-free survival also resulted in significantly increased median disease-free survival in patients receiving the combination therapy (2.7 vs. 1.5 years, P < 0.001).
Stage D 2 Prostatic Carcinoma
To study the effects of combination therapy in metastatic disease, 617 patients (311 leuprolide + Eulexin™, 306 leuprolide + placebo) with previously untreated advanced prostatic carcinoma were enrolled in a large multicentered, controlled clinical trial.
Three and one-half years after the study was initiated, median survival had been reached. The median actuarial survival time was 34.9 months for patients treated with leuprolide and Eulexin™ versus 27.9 months for patients treated with leuprolide alone. This 7 month increment represents a 25% improvement in overall survival time with the Eulexin™ therapy. Analysis of progression-free survival showed a 2.6 month improvement in patients who received leuprolide plus Eulexin™, a 19% increment over leuprolide and placebo.
HOW SUPPLIED
Eulexin™ capsules USP, 125 mg, are available as opaque, beige/beige capsules, imprinted "par/753" on the cap and body. They are available in bottle of 180 (NDC 80725-600-18).
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature]. Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).