Get your patient on Ivabradine - Ivabradine tablet, Film Coated (Ivabradine)
Ivabradine - Ivabradine tablet, Film Coated prescribing information
INDICATIONS AND USAGE
Ivabradine tablets are hyperpolarization-activated cyclic nucleotide-gated channel blocker indicated:
- To reduce the risk of hospitalization for worsening heart failure in adult patients with stable, symptomatic chronic heart failure with reduced left ventricular ejection fraction. (1.1 )
Heart Failure in Adult Patients
Ivabradine tablets are indicated to reduce the risk of hospitalization for worsening heart failure in adult patients with stable, symptomatic chronic heart failure with left ventricular ejection fraction ≤ 35%, who are in sinus rhythm with resting heart rate ≥ 70 beats per minute and either are on maximally tolerated doses of beta-blockers or have a contraindication to beta-blocker use.
DOSAGE AND ADMINISTRATION
Adult patients • Starting dose is 2.5 (vulnerable adults) or 5 mg twice daily with food. After 2 weeks of treatment, adjust dose based on heart rate. The maximum dose is 7.5 mg twice daily. (2.1 )
Adults
The recommended starting dose of ivabradine tablet is 5 mg twice daily with food. Assess patient after two weeks and adjust dose to achieve a resting heart rate between 50 and 60 beats per minute (bpm) as shown in Table 1. Thereafter, adjust dose as needed based on resting heart rate and tolerability. The maximum dose is 7.5 mg twice daily. In adult patients unable to swallow tablets, ivabradine oral solution can be used [see Clinical Pharmacology (12.3)] . In patients with a history of conduction defects or other patients in whom bradycardia could lead to hemodynamic compromise, initiate therapy at 2.5 mg twice daily before increasing the dose based on heart rate [see Warnings and Precautions (5.3)] .
Table 1. Dose Adjustment for Adults
| Heart Rate | Dose Adjustment |
| > 60 bpm | Increase dose by 2.5 mg (given twice daily) up to a maximum dose of 7.5 mg twice daily |
| 50 to 60 bpm | Maintain dose |
| < 50 bpm or signs and symptoms of bradycardia | Decrease dose by 2.5 mg (given twice daily); if current dose is 2.5 mg twice daily, discontinue therapy• |
• [see Warnings and Precautions (5.3)]
DOSAGE FORMS AND STRENGTHS
Ivabradine tablets 5 mg having functional scoring are light salmon to salmon-colored, oval-shaped, film-coated tablets scored on both edges, debossed with ‘A’, score and ‘7’ on one face and plain on the other face.
Ivabradine tablets 7.5 mg are light salmon to salmon-colored, triangular-shaped, film-coated tablets debossed with ‘662’ on one face and ‘L’ on the other face.
USE IN SPECIFIC POPULATIONS
- Lactation: Breastfeeding not recommended. (8.2 )
Pregnancy
Risk Summary
Based on findings in animals, ivabradine may cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies of ivabradine in pregnant women to inform any drug-associated risks. In animal reproduction studies, oral administration of ivabradine to pregnant rats during organogenesis at a dosage providing 1 to 3 times the human exposure (AUC 0-24hr ) at the MRHD resulted in embryo-fetal toxicity and teratogenicity manifested as abnormal shape of the heart, interventricular septal defect, and complex anomalies of primary arteries. Increased post-natal mortality was associated with these teratogenic effects in rats. In pregnant rabbits, increased post-implantation loss was noted at an exposure (AUC 0-24hr ) 5 times the human exposure at the MRHD. Lower doses were not tested in rabbits. The background risk of major birth defects for the indicated population is unknown. The estimated background risk of major birth defects in the U.S. general population is 2 to 4%, however, and the estimated risk of miscarriage is 15 to 20% in clinically recognized pregnancies. Advise a pregnant woman of the potential risk to the fetus.
Clinical Considerations
Disease-associated Maternal and/or Embryo-fetal Risk
Stroke volume and heart rate increase during pregnancy, increasing cardiac output, especially during the first trimester. Pregnant patients with left ventricular ejection fraction less than 35% on maximally tolerated doses of beta-blockers may be particularly heart rate dependent for augmenting cardiac output. Therefore, pregnant patients who are started on ivabradine, especially during the first trimester, should be followed closely for destabilization of their congestive heart failure that could result from heart rate slowing.
Monitor pregnant women with chronic heart failure in 3 rd trimester of pregnancy for preterm birth.
Data
Animal Data
In pregnant rats, oral administration of ivabradine during the period of organogenesis (gestation day 6 to 15) at doses of 2.3, 4.6, 9.3, or 19 mg/kg/day resulted in fetal toxicity and teratogenic effects. Increased intrauterine and post-natal mortality and cardiac malformations were observed at doses ≥ 2.3 mg/kg/day (equivalent to the human exposure at the MRHD based on AUC 0-24hr ). Teratogenic effects including interventricular septal defect and complex anomalies of major arteries were observed at doses ≥ 4.6 mg/kg/day (approximately 3 times the human exposure at the MRHD based on AUC 0-24hr ).
In pregnant rabbits, oral administration of ivabradine during the period of organogenesis (gestation day 6 to 18) at doses of 7, 14, or 28 mg/kg/day resulted in fetal toxicity and teratogenicity. Treatment with all doses ≥ 7 mg/kg/day (equivalent to the human exposure at the MRHD based on AUC 0-24hr ) caused an increase in post-implantation loss. At the high dose of 28 mg/kg/day (approximately 15 times the human exposure at the MRHD based on AUC 0-24hr ), reduced fetal and placental weights were observed, and evidence of teratogenicity (ectrodactylia observed in 2 of 148 fetuses from 2 of 18 litters) was demonstrated.
In the pre-and post-natal study, pregnant rats received oral administration of ivabradine at doses of 2.5, 7, or 20 mg/kg/day from gestation day 6 to lactation day 20. Increased post-natal mortality associated with cardiac teratogenic findings was observed in the F1 pups delivered by dams treated at the high dose (approximately 15 times the human exposure at the MRHD based on AUC 0-24hr ).
Lactation
Risk Summary
There is no information regarding the presence of ivabradine in human milk, the effects of ivabradine on the breastfed infant, or the effects of the drug on milk production. Animal studies have shown, however, that ivabradine is present in rat milk [see Data] . Because of the potential risk to breastfed infants from exposure to ivabradine, breastfeeding is not recommended.
Data
Lactating rats received daily oral doses of [14C]-ivabradine (7 mg/kg) on post-parturition days 10 to 14; milk and maternal plasma were collected at 0.5 and 2.5 hours post-dose on day 14. The ratios of total radioactivity associated with [14C]-ivabradine or its metabolites in milk vs. plasma were 1.5 and 1.8, respectively, indicating that ivabradine is transferred to milk after oral administration.
Females and Males of Reproductive Potential
Contraception
Females
Ivabradine may cause fetal harm, based on animal data. Advise females of reproductive potential to use effective contraception during ivabradine treatment [see Use in Specific Populations (8.1)] .
Pediatric Use
The safety and efficacy of ivabradine have not been established in patients less than 6 months of age.
Geriatric Use
No pharmacokinetic differences have been observed in elderly (≥ 65 years) or very elderly (≥ 75 years) patients compared to the overall population. However, ivabradine has only been studied in a limited number of patients ≥ 75 years of age.
Hepatic Impairment
No dose adjustment is required in patients with mild or moderate hepatic impairment. Ivabradine is contraindicated in patients with severe hepatic impairment (Child-Pugh C) as it has not been studied in this population and an increase in systemic exposure is anticipated [see Contraindications (4) and Clinical Pharmacology (12.3)] .
Renal Impairment
No dosage adjustment is required for patients with creatinine clearance 15 to 60 mL/min. No data are available for patients with creatinine clearance below 15 mL/min [see Clinical Pharmacology (12.3)] .
CONTRAINDICATIONS
Ivabradine tablets are contraindicated in patients with:
- Acute decompensated heart failure
- Clinically significant hypotension
- Sick sinus syndrome, sinoatrial block or 3 rd degree AV block, unless a functioning demand pacemaker is present
- Clinically significant bradycardia [see Warnings and Precautions (5.3)]
- Severe hepatic impairment [see Use in Specific Populations (8.6)]
- Pacemaker dependence (heart rate maintained exclusively by the pacemaker) [see Drug Interactions (7.3)]
- Concomitant use of strong cytochrome P450 3A4 (CYP3A4) inhibitors [see Drug Interactions (7.1)]
WARNINGS AND PRECAUTIONS
Fetal Toxicity
Ivabradine may cause fetal toxicity when administered to a pregnant woman based on findings in animal studies. Embryo-fetal toxicity and cardiac teratogenic effects were observed in fetuses of pregnant rats treated during organogenesis at exposures 1 to 3 times the human exposures (AUC 0-24hr ) at the maximum recommended human dose (MRHD) [see Use in Specific Populations (8.1)] . Advise females of reproductive potential to use effective contraception when taking ivabradine [see Use in Specific Populations (8.3)] .
Atrial Fibrillation
Ivabradine increases the risk of atrial fibrillation. In the Systolic Heart Failure Treatment with the I f Inhibitor Ivabradine Trial (SHIFT), the rate of atrial fibrillation was 5% per patient-year in patients treated with ivabradine and 3.9% per patient-year in patients treated with placebo [see Clinical Studies (14)] . Regularly monitor cardiac rhythm. Discontinue ivabradine if atrial fibrillation develops.
Bradycardia and Conduction Disturbances
Adult Patients
Bradycardia, sinus arrest, and heart block have occurred with ivabradine. The rate of bradycardia was 6% per patient-year in patients treated with ivabradine (2.7% symptomatic; 3.4% asymptomatic) and 1.3% per patient-year in patients treated with placebo. Risk factors for bradycardia include sinus node dysfunction, conduction defects (e.g., 1 st or 2 nd degree atrioventricular block, bundle branch block), ventricular dyssynchrony, and use of other negative chronotropes (e.g., digoxin, diltiazem, verapamil, amiodarone). Bradycardia may increase the risk of QT prolongation which may lead to severe ventricular arrhythmias, including torsade de pointes, especially in patients with risk factors such as use of QTc prolonging drugs [see Adverse Reactions (6.2)] .
Concurrent use of verapamil or diltiazem will increase ivabradine exposure, may themselves contribute to heart rate lowering, and should be avoided [see Clinical Pharmacology (12.3)] . Avoid use of ivabradine in patients with 2 nd degree atrioventricular block unless a functioning demand pacemaker is present [see Contraindications (4)] .
ADVERSE REACTIONS
Clinically significant adverse reactions that appear in other sections of the labeling include:
- Atrial Fibrillation [see Warnings and Precautions (5.2)]
- Bradycardia and Conduction Disturbances [see Warnings and Precautions (5.3)]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Patients with Heart Failure
In SHIFT, safety was evaluated in 3,260 patients treated with ivabradine and 3,278 patients given placebo. The median duration of ivabradine exposure was 21.5 months.
The most common adverse drug reactions in the SHIFT trial are shown in Table 2 [see Warnings and Precautions (5.2), (5.3)] .
Table 2. Adverse Drug Reactions with Rates ≥ 1% Higher on Ivabradine than Placebo occurring in > 1% on Ivabradine in SHIFT
| Ivabradine N = 3,260 | Placebo N = 3,278 | |
|---|---|---|
| Bradycardia | 10% | 2.2% |
| Hypertension, blood pressure increased | 8.9% | 7.8% |
| Atrial fibrillation | 8.3% | 6.6% |
| Phosphenes, visual brightness | 2.8% | 0.5% |
Luminous Phenomena (Phosphenes)
Phosphenes are phenomena described as a transiently enhanced brightness in a limited area of the visual field, halos, image decomposition (stroboscopic or kaleidoscopic effects), colored bright lights, or multiple images (retinal persistency). Phosphenes are usually triggered by sudden variations in light intensity. Ivabradine can cause phosphenes, thought to be mediated through ivabradine’s effects on retinal photoreceptors [see Clinical Pharmacology (12.1)] . Onset is generally within the first 2 months of treatment, after which they may occur repeatedly. Phosphenes were generally reported to be of mild to moderate intensity and led to treatment discontinuation in < 1% of patients; most resolved during or after treatment.
Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency reliably or establish a causal relationship to drug exposure.
The following adverse reactions have been identified in adults during post-approval use of ivabradine: syncope, hypotension, torsade de pointes, ventricular fibrillation, ventricular tachycardia, angioedema, erythema, rash, pruritus, urticaria, vertigo, and diplopia, and visual impairment.
DRUG INTERACTIONS
Cytochrome P450-Based Interactions
Ivabradine is primarily metabolized by CYP3A4. Concomitant use of CYP3A4 inhibitors increases ivabradine plasma concentrations and use of CYP3A4 inducers decreases them. Increased plasma concentrations may exacerbate bradycardia and conduction disturbances.
The concomitant use of strong CYP3A4 inhibitors is contraindicated [see Contraindications (4) and Clinical Pharmacology (12.3)] . Examples of strong CYP3A4 inhibitors include azole antifungals (e.g., itraconazole), macrolide antibiotics (e.g., clarithromycin, telithromycin), HIV protease inhibitors (e.g., nelfinavir), and nefazodone.
Avoid concomitant use of moderate CYP3A4 inhibitors when using ivabradine. Examples of moderate CYP3A4 inhibitors include diltiazem, verapamil, and grapefruit juice [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)] .
Avoid concomitant use of CYP3A4 inducers when using ivabradine. Examples of CYP3A4 inducers include St. John’s wort, rifampicin, barbiturates, and phenytoin [see Clinical Pharmacology (12.3)] .
Negative Chronotropes
Most patients receiving ivabradine will also be treated with a beta-blocker. The risk of bradycardia increases with concomitant administration of drugs that slow heart rate (e.g., digoxin, amiodarone, beta-blockers). Monitor heart rate in patients taking ivabradine with other negative chronotropes.
Pacemakers in Adults
Ivabradine dosing is based on heart rate reduction, targeting a heart rate of 50 to 60 beats per minute in adults [see Dosage and Administration (2.1)] . Patients with demand pacemakers set to a rate ≥ 60 beats per minute cannot achieve a target heart rate < 60 beats per minute, and these patients were excluded from clinical trials [see Clinical Studies (14.1)] . The use of ivabradine is not recommended in patients with demand pacemakers set to rates ≥ 60 beats per minute.
DESCRIPTION
Ivabradine tablets contain ivabradine as the active pharmaceutical ingredient. Ivabradine is a hyperpolarization-activated cyclic nucleotide-gated channel blocker that reduces the spontaneous pacemaker activity of the cardiac sinus node by selectively inhibiting the I f current, resulting in heart rate reduction with no effect on ventricular repolarization and no effects on myocardial contractility. The chemical name for ivabradine hydrochloride is 3-(3-{[((7S)-3,4-Dimethoxybicyclo[4.2.0]octa-1,3,5-trien-7-yl)methyl] methyl amino} propyl)-1,3,4,5-tetrahydro-7,8-dimethoxy-2H-3-benzazepin-2-one, hydrochloride. The molecular formula is C 27 H 36 N 2 O 5 ·HCl, and the molecular weight (free base + HCl) is 505.1 (468.6 + 36.5). The chemical structure of ivabradine is shown in Figure 1. Figure 1. Chemical Structure of Ivabradine 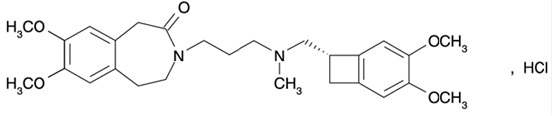 Ivabradine tablets are formulated as light salmon to salmon colored, film-coated tablets for oral administration in strengths of 5 mg and 7.5 mg of ivabradine, equivalent to 5.39 mg and 8.09 mg of ivabradine hydrochloride respectively. The tablets contain the following inactive ingredients: maltodextrin, lactose anhydrous, butylated hydroxyanisole, butylated hydroxytoluene, hypromellose 2910, colloidal silicon dioxide, magnesium stearate, hydroxypropyl cellulose, titanium dioxide, polyethylene glycol 6000, iron oxide yellow and iron oxide red.
Ivabradine tablets are formulated as light salmon to salmon colored, film-coated tablets for oral administration in strengths of 5 mg and 7.5 mg of ivabradine, equivalent to 5.39 mg and 8.09 mg of ivabradine hydrochloride respectively. The tablets contain the following inactive ingredients: maltodextrin, lactose anhydrous, butylated hydroxyanisole, butylated hydroxytoluene, hypromellose 2910, colloidal silicon dioxide, magnesium stearate, hydroxypropyl cellulose, titanium dioxide, polyethylene glycol 6000, iron oxide yellow and iron oxide red.
CLINICAL PHARMACOLOGY
Mechanism of Action
Ivabradine blocks the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel responsible for the cardiac pacemaker I f current, which regulates heart rate. In clinical electrophysiology studies, the cardiac effects were most pronounced in the sinoatrial (SA) node, but prolongation of the AH interval has occurred as has PR interval prolongation. There was no effect on ventricular repolarization and no effects on myocardial contractility [see Clinical Pharmacology (12.2)] .
Ivabradine can also inhibit the retinal current I h . I h is involved in curtailing retinal responses to bright light stimuli. Under triggering circumstances (e.g., rapid changes in luminosity), partial inhibition of I h by ivabradine may underlie the luminous phenomena experienced by patients. Luminous phenomena (phosphenes) are described as a transient enhanced brightness in a limited area of the visual field [see Adverse Reactions (6.1)] .
Pharmacodynamics
Ivabradine causes a dose-dependent reduction in heart rate. The size of the effect is dependent on the baseline heart rate (i.e., greater heart rate reduction occurs in patients with higher baseline heart rate). At recommended doses, heart rate reduction is approximately 10 bpm at rest and during exercise. Analysis of heart rate reduction vs. dose indicates a plateau effect at doses > 20 mg twice daily. In a study of patients with preexisting conduction system disease (first-or second-degree AV block or left or right bundle branch block) requiring electrophysiologic study, IV ivabradine (0.2 mg/kg) administration slowed the overall heart rate by approximately 15 bpm, increased the PR interval (29 msec), and increased the AH interval (27 msec). Ivabradine does not have negative inotropic effects. Ivabradine increases the uncorrected QT interval with heart rate slowing but does not cause rate-corrected prolongation of QT.
Pharmacokinetics
The peak concentration (C max ) and area under the plasma concentration time curve (AUC) are similar for ivabradine and S 18982 between oral solution and tablets for the same dose.
Absorption and Bioavailability
Following oral administration, peak plasma ivabradine concentrations are reached in approximately 1 hour under fasting conditions. The absolute oral bioavailability of ivabradine is approximately 40% because of first-pass elimination in the gut and liver.
Food delays absorption by approximately 1 hour and increases plasma exposure by 20% to 40%. Ivabradine should be taken with food [see Dosage and Administration (2)] .
Ivabradine is approximately 70% plasma protein bound, and the volume of distribution at steady state is approximately 100 L.
Metabolism and Excretion
The pharmacokinetics of ivabradine are linear over an oral dose range of 0.5 mg to 24 mg. Ivabradine is extensively metabolized in the liver and intestines by CYP3A4-mediated oxidation. The major metabolite is the N-desmethylated derivative (S 18982), which is equipotent to ivabradine and circulates at concentrations approximately 40% that of ivabradine. The N-desmethylated derivative is also metabolized by CYP3A4. Ivabradine plasma levels decline with a distribution half-life of 2 hours and an effective half-life of approximately 6 hours.
The total clearance of ivabradine is 24 L/h, and renal clearance is approximately 4.2 L/h, with ~ 4% of an oral dose excreted unchanged in urine. The excretion of metabolites occurs to a similar extent via feces and urine.
Drug Interactions
The effects of coadministered drugs (CYP3A4 inhibitors, substrates, inducers, and other concomitantly administered drugs) on the pharmacokinetics of ivabradine were studied in several single-and multiple-dose studies. Pharmacokinetic measures indicating the magnitude of these interactions are presented in Figure 2.
Figure 2. Impact of Coadministered Drugs on the Pharmacokinetics of Ivabradine
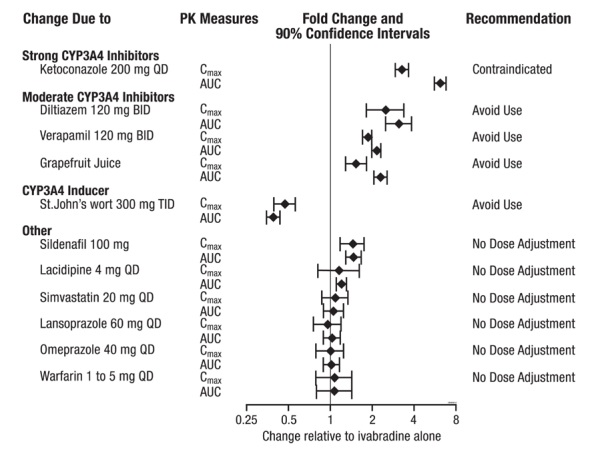
Digoxin exposure did not change when concomitantly administered with ivabradine. No dose adjustment is required when ivabradine is concomitantly administered with digoxin.
Effect of Ivabradine on Metformin Pharmacokinetics
Ivabradine, dosed at 10 mg twice daily to steady state, did not affect the pharmacokinetics of metformin (an organic cation transporter [OCT2] sensitive substrate). The geometric mean (90% confidence interval [CI]) ratios of C max and AUC inf of metformin, with and without ivabradine were 0.98 [0.83 to 1.15] and 1.02 [0.86 to 1.22], respectively. No dose adjustment is required for metformin when administered with ivabradine.
Specific Populations
Age
No pharmacokinetic differences (AUC or C max ) have been observed between elderly (≥ 65 years) or very elderly (≥ 75 years) patients and the overall patient population [see Use in Specific Populations (8.5)] .
Hepatic Impairment
In patients with mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment, the pharmacokinetics of ivabradine were similar to that in patients with normal hepatic function. No data are available in patients with severe hepatic impairment (Child-Pugh C) [see Contraindications (4)] .
Renal Impairment
Renal impairment (creatinine clearance from 15 to 60 mL/min) has minimal effect on the pharmacokinetics of ivabradine. No data are available for patients with creatinine clearance below 15 mL/min.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
There was no evidence of carcinogenicity when mice and rats received ivabradine up to 104 weeks by dietary administration. High doses in these studies were associated with mean ivabradine exposures of at least 37 times higher than the human exposure (AUC 0-24hr ) at the MRHD.
Ivabradine tested negative in the following assays: bacterial reverse mutation (Ames) assay, in vivo bone marrow micronucleus assay in both mouse and rat, in vivo chromosomal aberration assay in rats, and in vivo unscheduled DNA synthesis assay in rats. Results of the in vitro chromosomal aberration assay were equivocal at concentrations approximately 1,500 times the human C max at the MRHD. Ivabradine tested positive in the mouse lymphoma assays and in vitro unscheduled DNA synthesis assay in rat hepatocytes at concentrations greater than 1,500 times the human C max at the MRHD.
Reproduction toxicity studies in animals demonstrated that ivabradine did not affect fertility in male or female rats at exposures 46 to 133 times the human exposure (AUC 0-24hr ) at the MRHD.
Animal Toxicology and/or Pharmacology
Reversible changes in retinal function were observed in dogs administered oral ivabradine at total doses of 2, 7, or 24 mg/kg/day (approximately 0.6 to 50 times the human exposure at the MRHD based on AUC 0-24hr ) for 52 weeks. Retinal function assessed by electroretinography demonstrated reductions in cone system responses, which reversed within a week post-dosing, and were not associated with damage to ocular structures as evaluated by light microscopy. These data are consistent with the pharmacological effect of ivabradine related to its interaction with hyperpolarization-activated I h currents in the retina, which share homology with the cardiac pacemaker I f current.
CLINICAL STUDIES
Heart Failure in Adult Patients
SHIFT
The Systolic Heart Failure Treatment with the I f Inhibitor Ivabradine Trial (SHIFT) was a randomized, double-blind trial comparing ivabradine and placebo in 6,558 adult patients with stable New York Heart Association (NYHA) class II to IV heart failure, left ventricular ejection fraction ≤ 35%, and resting heart rate ≥ 70 bpm. Patients had to have been clinically stable for at least 4 weeks on an optimized and stable clinical regimen, which included maximally tolerated doses of beta-blockers and, in most cases, ACE inhibitors or ARBs, spironolactone, and diuretics, with fluid retention and symptoms of congestion minimized. Patients had to have been hospitalized for heart failure within 12 months prior to study entry.
The underlying cause of CHF was coronary artery disease in 68% of patients. At baseline, approximately 49% of randomized patients were NYHA class II, 50% were NYHA class III, and 2% were NYHA class IV. The mean left ventricular ejection fraction was 29%. All patients were initiated on ivabradine 5 mg (or matching placebo) twice daily and the dose was increased to 7.5 mg twice daily or decreased to 2.5 mg twice daily to maintain the resting heart rate between 50 and 60 bpm, as tolerated. The primary endpoint was a composite of the first occurrence of either hospitalization for worsening heart failure or cardiovascular death.
Most patients (89%) were taking beta-blockers, with 26% on guideline-defined target daily doses. The main reasons for not receiving the target beta-blocker doses at baseline were hypotension (45% of patients not at target), fatigue (32%), dyspnea (14%), dizziness (12%), history of cardiac decompensation (9%), and bradycardia (6%). For the 11% of patients not receiving any beta-blocker at baseline, the main reasons were chronic obstructive pulmonary disease, hypotension, and asthma. Most patients were also taking ACE inhibitors and/or angiotensin II antagonists (91%), diuretics (83%), and anti-aldosterone agents (60%). Few patients had an implantable cardioverter-defibrillator (ICD) (3.2%) or a cardiac resynchronization therapy (CRT) device (1.1%). Median follow-up was 22.9 months. At 1 month, 63%, 26%, and 8% of ivabradine -treated patients were taking 7.5, 5, and 2.5 mg BID, whereas 3% had withdrawn from the drug, primarily for bradycardia.
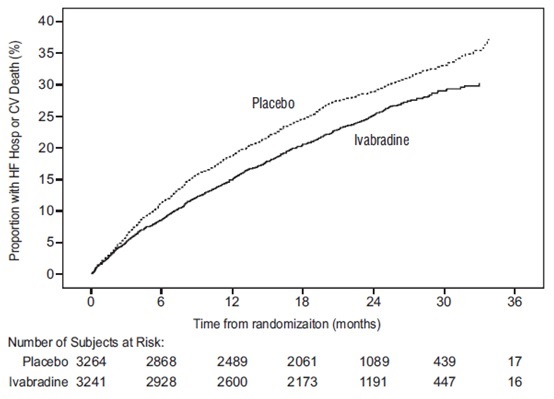
SHIFT demonstrated that ivabradine reduced the risk of the combined endpoint of hospitalization for worsening heart failure or cardiovascular death based on a time-to-event analysis (hazard ratio: 0.82, 95% confidence interval [CI]: 0.75, 0.9, p < 0.0001) (Table 3). The treatment effect reflected only a reduction in the risk of hospitalization for worsening heart failure; there was no favorable effect on the mortality component of the primary endpoint. In the overall treatment population, ivabradine had no statistically significant benefit on cardiovascular death.
Table 3. SHIFT – Incidence of the Primary Composite Endpoint and Components
| Endpoint | Ivabradine (N = 3,241) | Placebo (N = 3,264) | |||||||
| n | % | % PY | n | % | % PY | ||||
| Hazard Ratio | [95% CI] | p-value | |||||||
| Primary composite endpoint: Time to first hospitalization for worsening heart failure or cardiovascular death a | 793 | 24.5 | 14.5 | 937 | 28.7 | 17.7 | 0.82 | [0.75, 0.9] | < 0.0001 |
| Hospitalization for worsening heart failure | 505 | 15.6 | 9.2 | 660 | 20.2 | 12.5 | |||
| Cardiovascular death as first event | 288 | 8.9 | 4.8 | 277 | 8.5 | 4.7 | |||
| Patients with events at any time | |||||||||
| Hospitalization for worsening heart failure b | 514 | 15.9 | 9.4 | 672 | 20.6 | 12.7 | 0.74 | [0.66, 0.83] | |
| Cardiovascular death b | 449 | 13.9 | 7.5 | 491 | 15 | 8.3 | 0.91 | [0.8, 1.03] | |
a Patients who died on the same calendar day as their first hospitalization for worsening heart failure are counted under cardiovascular death.
b Analyses of the components of the primary composite endpoint were not prospectively planned to be adjusted for multiplicity.
N: number of patients at risk; n: number of patients having experienced the endpoint; %: incidence rate = (n/N) × 100; % PY: annual incidence rate = (n/number of patient-years) × 100; CI: confidence interval The hazard ratio between treatment groups (ivabradine/placebo) was estimated based on an adjusted Cox proportional hazards model with beta-blocker intake at randomization (yes/no) as a covariate; p-value: Wald test The Kaplan-Meier curve (Figure 3) shows time to first occurrence of the primary composite endpoint of hospitalization for worsening heart failure or cardiovascular death in the overall study.
Figure 3. SHIFT: Time to First Event of Primary Composite Endpoint
A wide range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes. Many of these results are shown in Figure 4. Such analyses must be interpreted cautiously, as differences can reflect the play of chance among a large number of analyses.
Most of the results show effects consistent with the overall study result. Ivabradine’s benefit on the primary endpoint in SHIFT appeared to decrease as the dose of beta-blockers increased, with little if any benefit demonstrated in patients taking guideline-defined target doses of beta-blockers. Figure 4. Effect of Treatment on Primary Composite Endpoint in Subgroups
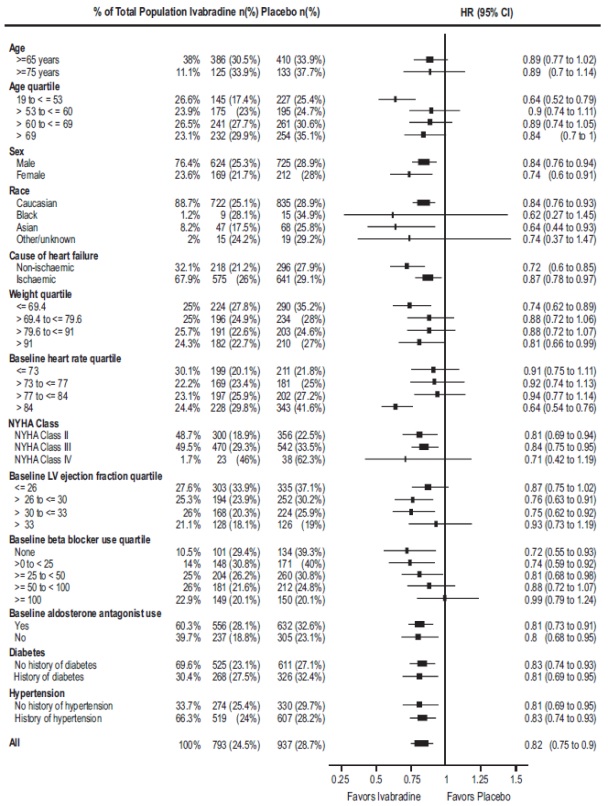
Note: The figure above presents effects in various subgroups, all of which are baseline characteristics.
The 95% confidence limits that are shown do not take into account the number of comparisons made and may not reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
BEAUTIFUL and SIGNIFY: No benefit in stable coronary artery disease with or without stable heart failure
The Morbidity-mortality Evaluation of the I f Inhibitor Ivabradine in Patients with Coronary Disease and Left Ventricular Dysfunction Trial (BEAUTIFUL) was a randomized, double-blind, placebo-controlled trial in 10,917 adult patients with coronary artery disease, impaired left ventricular systolic function (ejection fraction < 40%) and resting heart rate ≥ 60 bpm. Patients had stable symptoms of heart failure and/or angina for at least 3 months and were receiving conventional cardiovascular medications at stable doses for at least 1 month. Beta-blockertherapy was not required, nor was there a protocol mandate to achieve any specific dosing targets for patients who were taking beta-blockers. Patients were randomized 1:1 to ivabradine or placebo at an initial dose of 5 mg twice daily with the dose increased to 7.5 mg twice daily depending on resting heart rate and tolerability. The primary endpoint was the composite of time to first cardiovascular death, hospitalization for acute myocardial infarction, or hospitalization for new-onset or worsening heart failure. Most patients were NYHA class II (61.4%) or class III (23.2%) -none were class IV. Through a median follow-up of 19 months, ivabradine did not significantly affect the primary composite endpoint (HR 1, 95% CI = 0.91, 1.1).
The Study Assessing the Morbi-mortality Benefits of the I f Inhibitor Ivabradine in Patients with Coronary Artery Disease Trial (SIGNIFY) was a randomized, double-blind trial administering ivabradine or placebo to 19,102 adult patients with stable coronary artery disease but without clinically evident heart failure (NYHA class I). Beta-blocker therapy was not required. Ivabradine was initiated at a dose of 7.5 mg twice daily and the dose could be increased to as high as 10 mg twice daily or down-titrated to 5.0 mg twice daily to achieve a target heart rate of 55 to 60 bpm. The primary endpoint was a composite of the first occurrence of either cardiovascular death or myocardial infarction. Through a median follow-up of 24.1 months, ivabradine did not significantly affect the primary composite endpoint (HR 1.08, 95% CI = 0.96, 1.2).
HOW SUPPLIED/STORAGE AND HANDLING
Ivabradine tablets 5 mg having functional scoring are light salmon to salmon-colored, oval-shaped, film-coated tablets scored on both edges, debossed with ‘A’, score and ‘7’ on one face and plain on the other face. They are supplied as follows:
Bottle of 60 tablets with child resistant closure, NDC 46708-679-60
Bottle of 180 tablets with child resistant closure, NDC 46708-679-45
Ivabradine tablets 7.5 mg are light salmon to salmon-colored, triangular-shaped, film-coated tablets debossed with ‘662’ on one face and ‘L’ on the other face. They are supplied as follows:
Bottle of 60 tablets with child resistant closure, NDC 46708-680-60
Bottle of 180 tablets with child resistant closure, NDC 46708-680-45
Storage
Store ivabradine tablets at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Mechanism of Action
Ivabradine blocks the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel responsible for the cardiac pacemaker I f current, which regulates heart rate. In clinical electrophysiology studies, the cardiac effects were most pronounced in the sinoatrial (SA) node, but prolongation of the AH interval has occurred as has PR interval prolongation. There was no effect on ventricular repolarization and no effects on myocardial contractility [see Clinical Pharmacology (12.2)] .
Ivabradine can also inhibit the retinal current I h . I h is involved in curtailing retinal responses to bright light stimuli. Under triggering circumstances (e.g., rapid changes in luminosity), partial inhibition of I h by ivabradine may underlie the luminous phenomena experienced by patients. Luminous phenomena (phosphenes) are described as a transient enhanced brightness in a limited area of the visual field [see Adverse Reactions (6.1)] .