Get your patient on Posaconazole - Posaconazole tablet, Coated (Posaconazole)
Posaconazole - Posaconazole tablet, Coated prescribing information
INDICATIONS AND USAGE
Posaconazole is an azole antifungal indicated as follows:
- Posaconazole delayed-release tablets are indicated for the treatment of invasive aspergillosis in adults and pediatric patients 2 years of age and older who weigh greater than 40 kg. (1.1 )
- Posaconazole is indicated for the prophylaxis of invasive Aspergillus and Candida infections in patients who are at high risk of developing these infections due to being severely immunocompromised, such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD) or those with hematologic malignancies with prolonged neutropenia from chemotherapy as follows: (1.2 )
- Posaconazole delayed-release tablets : adults and pediatric patients 2 years of age and older who weigh greater than 40 kg
- Posaconazole oral suspension: adults and pediatric patients 13 years of age and older
- Posaconazole oral suspension is indicated for the treatment of oropharyngeal candidiasis (OPC), including OPC refractory (rOPC) to itraconazole and/or fluconazole in adults and pediatric patients 13 years of age and older. (1.3 )
1.1 Treatment of Invasive Aspergillosis
Posaconazole delayed-release tablets are indicated for the treatment of invasive aspergillosis in adults and pediatric patients 2 years of age and older who weigh greater than 40 kg.
Prophylaxis of Invasive Aspergillus and Candida Infections
Posaconazole is indicated for the prophylaxis of invasive Aspergillus and Candida infections in patients who are at high risk of developing these infections due to being severely immunocompromised, such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD) or those with hematologic malignancies with prolonged neutropenia from chemotherapy [see Clinical Studies (14.1) ] as follows:
- Posaconazole delayed-release tablets : adults and pediatric patients 2 years of age and older who weigh greater than 40 kg
- Posaconazole oral suspension: adults and pediatric patients 13 years of age and older
Treatment of Oropharyngeal Candidiasis Including Oropharyngeal Candidiasis Refractory to Itraconazole and/or Fluconazole
Posaconazole oral suspension is indicated for the treatment of oropharyngeal candidiasis, including oropharyngeal candidiasis refractory to itraconazole and/or fluconazole in adults and pediatric patients 13 years of age and older.
DOSAGE AND ADMINISTRATION
- Posaconazole formulations are supplied in different dose strengths of posaconazole, are approved for different indications, age groups, and weights, have different dosages and duration of therapy; and have different preparation and administration instructions. (2.1 )
- Posaconazole oral suspension is not substitutable with Posaconazole delayed-release tablets or Noxafil PowderMix for delayed-release oral suspension due to the differences in the dosing of each formulation. Therefore, follow the specific dosage recommendations for each of the formulations. (2.1 , 2.2 , 2.3 )
- Administer Posaconazole delayed-release tablets with or without food. (2.1 )
- Administer Posaconazole oral suspension with a full meal. (2.1 )
- See the full prescribing information for important administration and preparation instructions for Posaconazole (delayed-release tablets and Posaconazole oral suspension (2.5 , 2.6 , 2.7 )
- For adult and pediatric patients aged 2 years of age and older, see the Full Prescribing Information for dosing recommendations for Posaconazole delayed-release tablets and Posaconazole oral suspension based on the indication, age, and weight associated with the dosage form. (1.1 , 1.2 , 1.3 , 2.1 , 2.2 , 2.3 , 2.4 )
2.1 Important Administration Instructions
Posaconazole delayed-release tablets and Posaconazole oral suspension are approved for different indications, age groups and weights; have different dosages and duration of therapy; and have different preparation and administration instructions.
Therefore, select the recommended dosage form based on the indication, age group, and weight and carefully follow the recommended dosage, preparation and administration instructions described for each product [see Dosage and Administration (2.2 to 2.8) ] , and the following important administration instructions described below.
Non-substitutable
Posaconazole oral suspension is not substitutable with Posaconazole delayed-release tablets or Noxafil PowderMix for delayed-release oral suspension due to the differences in the dosing of each formulation. Therefore, follow the specific dosage recommendations for each of the formulations [see Dosage and Administration (2.2 , 2.3) ].
Posaconazole delayed-release tablets
- Swallow tablets whole. Do not divide, crush, or chew.
- Administer with or without food [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3) ] .
- For patients who cannot eat a full meal, Posaconazole delayed-release tablets should be used instead of Posaconazole oral suspension for the prophylaxis indication. Posaconazole delayed-release tablets generally provide higher plasma drug exposures than Posaconazole oral suspension under both fed and fasted conditions [see Dosage and Administration (2.6) ] .
Posaconazole oral suspension
- Administer with a full meal or with a liquid nutritional supplement or an acidic carbonated beverage (e.g., ginger ale) in patients who cannot eat a full meal [see Dosage and Administration (2.8) ].
2.2 Recommended Dosage of Posaconazole in Adult Patients
The recommended dosage of Posaconazole delayed-release tablets and Posaconazole oral suspension in adult patients for the treatment of invasive aspergillosis, prophylaxis of invasive Aspergillus and Candida infections in patients who are at high risk of developing these infections due to being severely immunocompromised, or for the treatment of oropharyngeal candidiasis (OPC) is shown in Table 1 [see Dosage and Administration (2.5 , 2.6 , 2.7) and Clinical Pharmacology (12.3) ].
| Dosage | Duration of Therapy |
|---|---|
| Treatment of Invasive Aspergillosis Switching between the Noxafil injection and Posaconazole delayed-release tablets is acceptable. A loading dose is not required when switching between dosage forms. | |
| Posaconazole Delayed-Release Tablets: Loading dose: 300 mg (three 100 mg delayed-release tablets) twice a day on the first day. Maintenance dose : 300 mg (three 100 mg delayed-release tablets) once a day, starting on the second day. | Loading dose: 1 day Maintenance dose: Recommended total duration of therapy is 6 to 12 weeks. |
| Prophylaxis of Invasive Aspergillus and Candida Infections | |
| Posaconazole Delayed-Release Tablets: Loading dose : 300 mg (three 100 mg delayed-release tablets) twice a day on the first day. Maintenance dose : 300 mg (three 100 mg delayed-release tablets) once a day, starting on the second day. Posaconazole Oral Suspension: 200 mg (5 mL) three times a day. | Loading dose : 1 day Maintenance dose : Duration of therapy is based on recovery from neutropenia or immunosuppression |
| Oropharyngeal Candidiasis (OPC) | |
| Posaconazole Oral Suspension: Loading dose : 100 mg (2.5 mL) twice a day on the first day. Maintenance dose : 100 mg (2.5 mL) once a day thereafter. | Loading dose : 1 day Maintenance dose : 13 days |
| OPC Refractory (rOPC) to Itraconazole and/or Fluconazole | |
| Posaconazole Oral Suspension: 400 mg (10 mL) twice a day. | Duration of therapy is based on the severity of the patient’s underlying disease and clinical response. |
2.3 Recommended Dosage of Posaconazole for the Treatment of Invasive Aspergillosis and Prophylaxis of Invasive Aspergillus and Candida Infections in Pediatric Patients 2 Years of Age and Older
Posaconazole delayed-release tablets
The recommended dosage of Posaconazole delayed-release tablets in pediatric patients 2 years of age and older who weigh greater than 40 kg for the treatment of invasive aspergillosis and prophylaxis of invasive Aspergillus and Candida infections is shown in Table 2 [see Dosage and Administration (2.5 , 2.6 , 2.7) and Clinical Pharmacology (12.3) ].
Posaconazole delayed-release tablets are not recommended for use in pediatric patients who weigh 40 kg or less because the recommended dosage cannot be achieved with this dosage form.
| Recommended Pediatric Dosage of Posaconazole Delayed-Release Tablets | Duration of Therapy |
|---|---|
| Posaconazole Delayed-Release Tablets (patients weighing greater than 40 kg): Loading dose: 300 mg (three 100 mg delayed-release tablets) twice a day on the first day. Maintenance dose: 300 mg (three 100 mg delayed-release tablets) once a day, starting on the second day. | Treatment of invasive aspergillosis: Recommended total duration of therapy is 6 to 12 weeks Prophylaxis of invasive Aspergillus and Candida infections: Duration of therapy is based on recovery from neutropenia or immunosuppression. |
Posaconazole Oral Suspension
The recommended dosage of Posaconazole oral suspension in pediatric patients 13 years of age and older for the prophylaxis of invasive Aspergillus and Candida Infections is shown in Table 3.
| Recommended Pediatric Dosage of Posaconazole Oral Suspension | Duration of Therapy |
|---|---|
| 200 mg (5 mL) three times a day | Duration of therapy is based on recovery from neutropenia or immunosuppression. |
2.4 Recommended Dosage of Posaconazole Oral Suspension for the Treatment of Oropharyngeal Candidiasis in Pediatric Patients 13 Years of Age and Older
The recommended dosage of Posaconazole oral suspension for the treatment of oropharyngeal candidiasis (OPC) and OPC refractory (rOPC) to itraconazole and/or fluconazole in pediatric patients 13 years of age and older is shown in Table 4.
The Posaconazole delayed-release tablets are not approved for the treatment of oropharyngeal candidiasis in pediatric patients.
| Recommended Pediatric Dosage of Posaconazole Oral Suspension | Duration of Therapy |
|---|---|
| Oropharyngeal Candidiasis (OPC) | |
| Loading Dose: 100 mg (2.5 mL) twice daily on the first day Maintenance Dose: 100 mg (2.5 mL) once daily | Loading dose: 1 day Maintenance dose: 13 days |
| OPC Refractory (rOPC) to Itraconazole and/or Fluconazole | |
| 400 mg (10 mL) twice daily | Duration of therapy is based on the severity of the patient’s underlying disease and clinical response. |
Administration Instructions for Posaconazole Delayed-Release Tablets
- Swallow the Posaconazole delayed-release tablets whole. Do not divide, crush, or chew.
- Administer Posaconazole delayed-release orally tablets with or without food [see Clinical Pharmacology (12.3) ].
2.6 Administration Instructions for Posaconazole Oral Suspension
Administer Posaconazole oral suspension with a full meal or with a liquid nutritional supplement or an acidic carbonated beverage (e.g., ginger ale) in patients who cannot eat a full meal .
For patients who cannot eat a full meal, use Posaconazole delayed-release tablets instead of the Posaconazole oral suspension for the prophylaxis of invasive Aspergillus and Candida infections in those who are at high risk of developing these infections due to being severely immunocompromised. This is because Posaconazole delayed-release tablets provide higher plasma drug exposures than Posaconazole oral suspension under fasted condition [see Dosage and Administration (2.1) ] .
For those patients using the Posaconazole oral suspension:
- Shake Posaconazole oral suspension well before use. Administer with measured dosing spoon (see Figure 1 ) provided.
- Administer with measured dosing spoon provided in the package (see Figure 1 ).
 |
| Figure 1: Measured dosing spoon provided in the package marked for doses of 2.5 mL and 5 mL. |
- Administer each dose of Posaconazole oral suspension during or immediately (i.e., within 20 minutes) following a full meal [see Clinical Pharmacology (12.3) ].
- In patients who cannot eat a full meal and for whom Posaconazole delayed-release tablets are not an option, administer each dose of Posaconazole oral suspension with a liquid nutritional supplement or an acidic carbonated beverage (e.g., ginger ale). If these patients cannot tolerate an oral nutritional supplement or an acidic carbonated beverage either use:
- an alternative antifungal therapy, or
- Posaconazole oral suspension and closely monitor patients for breakthrough fungal infections.
- Rinse the spoon with water after each administration and before storage.
Non-substitutability between Posaconazole Oral Suspension and Other Formulations
Posaconazole oral suspension is not substitutable with Posaconazole delayed-release tablets or Noxafil PowderMix for delayed-release oral suspension due to the differences in the dosing of each formulation. Therefore, follow the specific dosage recommendations for each of the formulations [see Dosage and Administration (2.2 , 2.3) ].
2.8 Dosage Modifications in Patients with Renal Impairment
The recommended dosage of Posaconazole oral suspension and Posaconazole delayed-release tablets is the same in patients with renal impairment compared to those with normal renal function.
DOSAGE FORMS AND STRENGTHS
Posaconazole Delayed-Release Tablets
100 mg of posaconazole: Yellow, coated, oblong tablets, debossed with "100" on one side.
Posaconazole Oral Suspension
4,200 mg/105 mL (40 mg/mL) of posaconazole: White, cherry-flavored suspension in amber glass bottles with child-resistant closures.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
Based on findings from animal data, posaconazole may cause fetal harm when administered to pregnant women. Available data for use of Noxafil in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, skeletal malformations were observed when posaconazole was dosed orally to pregnant rats during organogenesis at doses ≥1.4 times the 400 mg twice daily oral suspension regimen based on steady-state plasma concentrations of Noxafil in healthy volunteers. In pregnant rabbits dosed orally during organogenesis, doses of ≥3 times the clinical exposure caused an increase in resorptions (see Data ) . Based on animal data, advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data : Posaconazole resulted in maternal toxicity (reduced food consumption and reduced body weight gain) and skeletal malformations (cranial malformations and missing ribs) when given orally to pregnant rats during organogenesis (Gestational Days 6 through 15) at doses ≥27 mg/kg (≥1.4 times the 400 mg twice daily oral suspension regimen based on steady-state plasma concentrations of drug in healthy volunteers). The no-effect dose for malformations and maternal toxicity in rats was 9 mg/kg, which is 0.7 times the exposure achieved with the 400 mg twice daily oral suspension regimen. No malformations were seen in rabbits dosed during organogenesis (Gestational Days 7 through 19) at doses up to 80 mg/kg (5 times the exposure achieved with the 400 mg twice daily oral suspension regimen). In the rabbit, the no-effect dose was 20 mg/kg, while high doses of 40 mg/kg and 80 mg/kg (3 or 5 times the clinical exposure) caused an increase in resorptions. In rabbits dosed at 80 mg/kg, a reduction in body weight gain of females and a reduction in litter size were seen.
Lactation
Risk Summary
There are no data on the presence of posaconazole in human milk, the effects on the breastfed infant, or the effects on milk production. Posaconazole is excreted in the milk of lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for posaconazole and any potential adverse effects on the breastfed child from posaconazole or from the underlying maternal condition.
Pediatric Use
The posaconazole dosage forms (delayed-release tablets and oral suspension) are different products; are approved for different pediatric indications, age groups, and weights; have different dosing regimens; and have different preparation and administration instructions. Therefore, select the recommended dosage form based on the pediatric indication, age group, and weight [see Dosage and Administration (2.1) ].
Treatment of Invasive Aspergillosis
The safety and effectiveness of Posaconazole delayed-release tablets have been established for the treatment of invasive aspergillosis in pediatric patients 2 years of age and older.
Use of posaconazole for these pediatric indications is supported by evidence from adequate and well-controlled studies of Noxafil in adults and safety and pharmacokinetic (PK) data from pediatric studies [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3) ] . The safety of Noxafil in pediatric patients for these pediatric indications was consistent with the known safety profile of Noxafil in adults [see Adverse Reactions (6.1) ].
The safety and effectiveness of posaconazole have not been established in pediatric patients less than 2 years of age .
Prophylaxis of Invasive Aspergillus and Candida Infections
The safety and effectiveness of Posaconazole delayed-release tablets have been established for the prophylaxis of invasive Aspergillus and Candida infections in pediatric patients 2 years of age and older who are at high risk of developing these infections due to being severely immunocompromised.
The safety and effectiveness of Posaconazole oral suspension have been established for the prophylaxis of invasive Aspergillus and Candida infections in pediatric patients 13 years of age and older who are at high risk of developing these infections due to being severely immunocompromised.
Use of posaconazole for these pediatric indications is supported by adequate and well-controlled studies of Noxafil in adults and pediatric patients aged 13 years and older and additional PK and safety data in pediatric patients 2 years of age and older [see Clinical Pharmacology (12.3) and Clinical Studies (14) ] .
The safety and effectiveness of posaconazole have not been established in pediatric patients younger than 2 years of age.
Treatment of Oropharyngeal Candidiasis, including Refractory to Itraconazole and/or Fluconazole
The safety and effectiveness of Posaconazole oral suspension have been established for the treatment of oropharyngeal candidiasis (OPC), including OPC refractory (rOPC) to itraconazole and/or fluconazole in pediatric patients 13 years of age and older.
Use of Posaconazole oral suspension for this pediatric indication is supported by adequate and well controlled studies in adults and pediatric patients 13 years of age and older [see Clinical studies (14.4) ].
Posaconazole delayed-release tablets are not approved for the treatment of oropharyngeal candidiasis in pediatric patients. Posaconazole oral suspension is the only dosage form approved for the treatment of OPC and rOPC in pediatric patients [see Dosage and Administration (2.4) ].
The safety and effectiveness of Posaconazole oral suspension for the treatment of OPC and rOPC have not been established in pediatric patients less than 13 years of age.
Geriatric Use
No overall differences in the safety of Noxafil delayed-release tablets and Noxafil oral suspension have been observed between geriatric patients and younger adult patients in the clinical trials; therefore, the recommended dosage in geriatric patients is the same as that for younger adult patients. No clinically meaningful differences in posaconazole pharmacokinetics were observed in Noxafil-treated geriatric patients compared to Noxafil-treated younger adult patients during clinical trials [see Clinical Pharmacology (12.3) ] .
- Of the 230 patients treated with Noxafil delayed-release tablets, 38 (17%) patients were >65 years of age.
- Of the 605 patients treated with Noxafil oral suspension in Noxafil Oral Suspension Study 1 and Study 2 (prophylaxis of invasive Aspergillus and Candida infections in those at high risk of developing these infections due to being severely immunocompromised), 63 (10%) patients were ≥65 years of age.
- In studies of Noxafil for an unapproved indication, 48 patients treated with Noxafil oral suspension (greater than or equal to 800 mg/day (eight times the maximum recommended maintenance dosage for the treatment of OPC)) were ≥65 years of age.
- Of the 288 patients treated with Noxafil injection or Noxafil delayed-release tablets in the Aspergillosis Treatment Study, 85 (29%) patients were ≥65 years of age.
Renal Impairment
Posaconazole Oral Suspension and Posaconazole Delayed-Release Tablets
No dosage adjustment is required for patients with eGFR 20 mL/minute/1.73 m 2 or higher.
Due to variability in posaconazole exposure, closely monitor patients with eGFR less than 20 mL/minute/1.73 m 2 for breakthrough fungal infections. [see Clinical Pharmacology (12.3) ] .
Hepatic Impairment
No dose adjustment is recommended of posaconazole in patients with mild, moderate, or severe hepatic impairment (Child-Pugh Class A, B, or C, respectively) [see Pharmacology (12.3) ] . However, a specific hepatic impairment study has not been conducted with the Posaconazole delayed-release tablets.
Sex
No adjustment in the dosage of posaconazole is necessary based on sex.
Race
No adjustment in the dosage of posaconazole is necessary based on race.
Weight
Pharmacokinetic modeling suggests that patients who weigh greater than 120 kg may have lower posaconazole plasma drug exposure. Therefore, consider closely monitoring for breakthrough fungal infections particularly when using Posaconazole oral suspension in patients weighing greater than 120 kg [see Clinical Pharmacology (12.3) ] .
CONTRAINDICATIONS
- Known hypersensitivity to posaconazole or other azole antifungal agents. (4.1 )
- Coadministration of posaconazole with the following drugs is contraindicated: posaconazole increases concentrations and toxicities of:
- Sirolimus (4.2 , 7.2 )
- CYP3A4 substrates (pimozide, quinidine): can result in QTc interval prolongation and cases of torsades de pointes (TdP) (4.3 , 5.2 , 7.2 )
- HMG-CoA Reductase Inhibitors Primarily Metabolized through CYP3A4 (4.4 , 7.2 )
- Ergot alkaloids (4.5 , 7.2 )
- Venetoclax: In patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) at initiation and during the ramp-up phase (4.6 , 5.10 , 7.2 )
Hypersensitivity
Posaconazole is contraindicated in persons with known hypersensitivity to posaconazole or other azole antifungal agents.
Use with Sirolimus
Posaconazole is contraindicated with sirolimus. Concomitant administration of posaconazole with sirolimus increases the sirolimus blood concentrations by approximately 9-fold and can result in sirolimus toxicity [see Drug Interactions (7.2) and Clinical Pharmacology (12.3) ] .
QT Prolongation with Concomitant Use with CYP3A4 Substrates
Posaconazole is contraindicated with CYP3A4 substrates that prolong the QT interval. Concomitant administration of posaconazole with the CYP3A4 substrates, pimozide and quinidine may result in increased plasma concentrations of these drugs, leading to QTc prolongation and cases of torsades de pointes [see Warnings and Precautions (5.2) and Drug Interactions (7.2) ] .
HMG-CoA Reductase Inhibitors Primarily Metabolized Through CYP3A4
Coadministration with the HMG-CoA reductase inhibitors that are primarily metabolized through CYP3A4 (e.g., atorvastatin, lovastatin, and simvastatin) is contraindicated since increased plasma concentration of these drugs can lead to rhabdomyolysis [see Drug Interactions (7.3) and Clinical Pharmacology (12.3) ] .
Use with Ergot Alkaloids
Posaconazole may increase the plasma concentrations of ergot alkaloids (ergotamine and dihydroergotamine) which may lead to ergotism [see Drug Interactions (7.2) ] .
Use with Venetoclax
Coadministration of posaconazole with venetoclax at initiation and during the ramp-up phase is contraindicated in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) due to the potential for increased risk of tumor lysis syndrome [see Warnings and Precautions (5.10) and Drug Interactions (7.2) ].
WARNINGS AND PRECAUTIONS
- Calcineurin-Inhibitor Toxicity: Posaconazole increases concentrations of cyclosporine or tacrolimus; reduce dose of cyclosporine and tacrolimus and monitor concentrations frequently. (5.1 )
- Arrhythmias and QTc Prolongation: Posaconazole has been shown to prolong the QTc interval and cause cases of TdP. Administer with caution to patients with potentially proarrhythmic conditions. Do not administer with drugs known to prolong QTc interval and metabolized through CYP3A4. (5.2 , 7.2 )
- Electrolyte Disturbances: Monitor and correct, especially those involving potassium (K + ), magnesium (Mg ++ ), and calcium (Ca ++ ), before and during posaconazole therapy. (5.3 )
- Pseudoaldosteronism : Manifested by the onset or worsening of hypertension, and abnormal laboratory findings. Monitor blood pressure and potassium levels, and manage as necessary. (5.4 )
- Hepatic Toxicity: Elevations in liver tests may occur. Discontinuation should be considered in patients who develop abnormal liver tests or monitor liver tests during treatment. (5.5 )
- Concomitant Use with Midazolam: Posaconazole can prolong hypnotic/sedative effects. Monitor patients and benzodiazepine receptor antagonists should be available. (5.7 , 7.2 )
- Vincristine Toxicity: Concomitant administration of azole antifungals, including posaconazole, with vincristine has been associated with neurotoxicity and other serious adverse reactions; reserve azole antifungals, including posaconazole, for patients receiving a vinca alkaloid, including vincristine, who have no alternative antifungal treatment options. (5.8 , 7.2 )
- Breakthrough Fungal Infections : Monitor patients with severe diarrhea or vomiting when receiving Posaconazole delayed-release tablets and Posaconazole oral suspension. (5.9 )
- Venetoclax Toxicity: Concomitant administration of posaconazole with venetoclax may increase venetoclax toxicities, including the risk of tumor lysis syndrome, neutropenia, and serious infections; monitor for toxicity and reduce venetoclax dose. (4.6 , 5.10 , 7.2 )
Calcineurin-Inhibitor Toxicity
Concomitant administration of posaconazole with cyclosporine or tacrolimus increases the whole blood trough concentrations of these calcineurin-inhibitors [see Drug Interactions (7.2) and Clinical Pharmacology (12.3) ] . Nephrotoxicity and leukoencephalopathy (including deaths) have been reported in clinical efficacy studies in patients with elevated cyclosporine or tacrolimus concentrations. Frequent monitoring of tacrolimus or cyclosporine whole blood trough concentrations should be performed during and at discontinuation of posaconazole treatment and the tacrolimus or cyclosporine dose adjusted accordingly.
Arrhythmias and QT Prolongation
Some azoles, including posaconazole, have been associated with prolongation of the QT interval on the electrocardiogram. In addition, cases of torsades de pointes have been reported in patients taking posaconazole.
Results from a multiple time-matched ECG analysis in healthy volunteers did not show any increase in the mean of the QTc interval. Multiple, time-matched ECGs collected over a 12-hour period were recorded at baseline and steady-state from 173 healthy male and female volunteers (18-85 years of age) administered Noxafil oral suspension 400 mg twice daily with a high-fat meal. In this pooled analysis, the mean QTc (Fridericia) interval change from baseline was –5 msec following administration of the recommended clinical dose. A decrease in the QTc(F) interval (–3 msec) was also observed in a small number of subjects (n=16) administered placebo. The placebo-adjusted mean maximum QTc(F) interval change from baseline was <0 msec (–8 msec). No healthy subject administered Noxafil had a QTc(F) interval ≥500 msec or an increase ≥60 msec in their QTc(F) interval from baseline.
Posaconazole should be administered with caution to patients with potentially proarrhythmic conditions. Do not administer with drugs that are known to prolong the QTc interval and are metabolized through CYP3A4 [see Contraindications (4.3) and Drug Interactions (7.2) ] .
Electrolyte Disturbances
Electrolyte disturbances, especially those involving potassium, magnesium or calcium levels, should be monitored and corrected as necessary before and during posaconazole therapy.
Pseudoaldosteronism
Pseudoaldosteronism, manifested by the onset of hypertension or worsening of hypertension, and abnormal laboratory findings (hypokalemia, low serum renin and aldosterone, and elevated 11-deoxycortisol), has been reported with posaconazole use in the postmarket setting. Monitor blood pressure and potassium levels and manage as necessary. Management of pseudoaldosteronism may include discontinuation of posaconazole, substitution with an appropriate antifungal drug that is not associated with pseudoaldosteronism, or use of aldosterone receptor antagonists.
Hepatic Toxicity
Hepatic reactions (e.g., mild to moderate elevations in alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, total bilirubin, and/or clinical hepatitis) have been reported in clinical trials. The elevations in liver tests were generally reversible on discontinuation of therapy, and in some instances these tests normalized without drug interruption. Cases of more severe hepatic reactions including cholestasis or hepatic failure including deaths have been reported in patients with serious underlying medical conditions (e.g., hematologic malignancy) during treatment with posaconazole. These severe hepatic reactions were seen primarily in subjects receiving the Posaconazole oral suspension 800 mg daily (400 mg twice daily or 200 mg four times a day) in clinical trials.
Liver tests should be evaluated at the start of and during the course of posaconazole therapy. Patients who develop abnormal liver tests during posaconazole therapy should be monitored for the development of more severe hepatic injury. Patient management should include laboratory evaluation of hepatic function (particularly liver tests and bilirubin). Discontinuation of posaconazole must be considered if clinical signs and symptoms consistent with liver disease develop that may be attributable to posaconazole.
Renal Impairment
Due to the variability in exposure with Posaconazole delayed-release tablets and Posaconazole oral suspension, patients with severe renal impairment should be monitored closely for breakthrough fungal infections [see Dosage and Administration (2.4) and Use in Specific Populations (8.6) ].
Midazolam Toxicity
Concomitant administration of posaconazole with midazolam increases the midazolam plasma concentrations by approximately 5-fold. Increased plasma midazolam concentrations could potentiate and prolong hypnotic and sedative effects. Patients must be monitored closely for adverse effects associated with high plasma concentrations of midazolam and benzodiazepine receptor antagonists must be available to reverse these effects [see Drug Interactions (7.2) and Clinical Pharmacology (12.3) ] .
Vincristine Toxicity
Concomitant administration of azole antifungals, including posaconazole, with vincristine has been associated with neurotoxicity and other serious adverse reactions, including seizures, peripheral neuropathy, syndrome of inappropriate antidiuretic hormone secretion, and paralytic ileus. Reserve azole antifungals, including posaconazole, for patients receiving a vinca alkaloid, including vincristine, who have no alternative antifungal treatment options [see Drug Interactions (7.2) ] .
Breakthrough Fungal Infections
Patients who have severe diarrhea or vomiting should be monitored closely for breakthrough fungal infections when receiving Posaconazole delayed-release tablets or Posaconazole oral suspension.
Venetoclax Toxicity
Concomitant administration of posaconazole, a strong CYP3A4 inhibitor, with venetoclax may increase venetoclax toxicities, including the risk of tumor lysis syndrome (TLS), neutropenia, and serious infections. In patients with CLL/SLL, administration of posaconazole during initiation and the ramp-up phase of venetoclax is contraindicated [see Contraindications (4.6) ] . Refer to the venetoclax labeling for safety monitoring and dose reduction in the steady daily dosing phase in CLL/SLL patients.
For patients with acute myeloid leukemia (AML), dose reduction and safety monitoring are recommended across all dosing phases when coadministering posaconazole with venetoclax [see Drug Interactions (7.2) ]. Refer to the venetoclax prescribing information for dosing instructions.
ADVERSE REACTIONS
The following serious and otherwise important adverse reactions are discussed in detail in another section of the labeling:
- Arrhythmias and QT Prolongation [see Warnings and Precautions (5.2) ]
- Electrolyte Disturbances [see Warnings and Precautions (5.3) ]
- Pseudoaldosteronism [see Warnings and Precautions (5.4) ]
- Hepatic Toxicity [see Warnings and Precautions (5.5) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of Noxafil cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Treatment of Invasive Aspergillosis in Adults and Adolescents (Noxafil Injection and Noxafil Delayed-Release Tablets)
The safety of Noxafil injection and Noxafil delayed-release tablets was assessed in a randomized, double-blind, active-controlled clinical study of Noxafil injection and Noxafil delayed-release tablets versus voriconazole for treatment of invasive aspergillosis (Aspergillosis Treatment Study). A total of 575 adult and pediatric patients 14 years of age and older (288 in the Noxafil group, 287 in voriconazole group (voriconazole for injection or voriconazole tablets)) with proven, probable or possible invasive aspergillosis were included. The median duration of treatment was 67 days for Noxafil injection or Noxafil delayed-release tablet and 64 days for voriconazole (voriconazole for injection). In this study, with 55% to 60% of patients started intravenous treatment with Noxafil (Noxafil injection) or voriconazole (voriconazole for injection). The median duration of the first instance of intravenous treatment (before switching to oral treatment or discontinuing or completing study treatment) was 9 days for both groups. Table 5 presents adverse reactions reported at an incidence of ≥10% in either one of the treatment groups in the Aspergillosis Treatment Study.
Adverse reactions leading to treatment discontinuation were reported for 34% of patients. The most commonly reported adverse reactions (>2% of patients) leading to treatment discontinuation were septic shock, respiratory failure, and bronchopulmonary aspergillosis in the Noxafil group, and septic shock and acute myeloid leukemia in the voriconazole group. The most frequently reported adverse reactions in the Noxafil-treated group were pyrexia (28%), hypokalemia (28%), and nausea (23%).
| Adverse Reactions | Noxafil injection or Noxafil delayed-release tablets n=288 (%) | Voriconazole for injection or Voriconazole tablets n=287 (%) |
|---|---|---|
| Percentage of Patients Reporting any Adverse Reaction | 97.6 | 97.6 |
| Hypokalemia | 28.5 | 17.1 |
| Pyrexia | 28.1 | 25.1 |
| Nausea | 22.6 | 17.8 |
| Diarrhea | 18.1 | 18.1 |
| Vomiting | 18.1 | 13.6 |
| Alanine aminotransferase increased | 14.6 | 12.9 |
| Febrile neutropenia | 14.6 | 13.2 |
| Aspartate aminotransferase increased | 13.2 | 12.5 |
| Pneumonia | 12.5 | 9.1 |
| Headache | 12.2 | 8.7 |
| Constipation | 11.1 | 8.0 |
| Edema peripheral | 11.1 | 8.4 |
| Epistaxis | 11.1 | 5.9 |
| Cough | 10.4 | 8.4 |
| Abdominal pain | 10.1 | 8.4 |
| Hypomagnesemia | 10.1 | 6.3 |
Clinical Trial Experience with Noxafil Delayed-Release Tablets for Prophylaxis of Invasive Aspergillus and Candida Infections
The safety of Noxafil delayed-release tablets has been assessed in 230 patients in clinical trials. Patients were enrolled in a non-comparative pharmacokinetic and safety trial of Noxafil delayed-release tablets when given as antifungal prophylaxis (Noxafil Delayed-Release Tablet Study). Patients were immunocompromised with underlying conditions including hematological malignancy, neutropenia post-chemotherapy, GVHD, and post HSCT. This patient population was 62% male, had a mean age of 51 years (range: 19-78 years, 17% of patients were ≥65 years of age), and were 93% White and 16% Hispanic. Noxafil delayed-release tablets were given for a median duration of 28 days. In this study, 20 adult patients received 200 mg daily dose and 210 adult patients received 300 mg daily dosage (following twice daily dosing on Day 1 in each cohort). Table 6 presents adverse reactions (incidence of >10%) observed in patients treated with the Noxafil delayed-release tablets 300 mg daily dosage in the Noxafil Delayed-Release Tablet Study.
The most frequently reported adverse reactions (>25%) in patients treated with Noxafil delayed-release tablets 300 mg once daily were diarrhea, pyrexia, and nausea. The most common adverse reaction leading to discontinuation of Noxafil delayed-release tablets 300 mg once daily was nausea (2%).
| Adverse Reactions | Noxafil delayed-release tablet (300 mg) n=210 (%) |
|---|---|
| Percentage of Patients Reporting any Adverse Reaction | 99 |
| Diarrhea | 29 |
| Pyrexia | 28 |
| Nausea | 27 |
| Hypokalemia | 22 |
| Cough | 17 |
| Edema Peripheral | 16 |
| Rash | 16 |
| Epistaxis | 14 |
| Headache | 14 |
| Mucosal Inflammation | 14 |
| Thrombocytopenia | 14 |
| Vomiting | 13 |
| Abdominal Pain | 11 |
| Hypertension | 11 |
| Anemia | 10 |
| Asthenia | 10 |
| Chills | 10 |
| Constipation | 10 |
| Hypomagnesemia | 10 |
Clinical Trials Safety Experience with Noxafil Oral Suspension
The safety of Noxafil oral suspension has been assessed in 1,844 patients, including:
- 605 patients in the active-controlled prophylaxis studies for the prophylaxis of invasive Aspergillus and Candida infections
- 557 patients in the active-controlled OPC studies (not refractory to itraconazole or fluconazole)
- 239 patients in refractory OPC studies (refractory to itraconazole or fluconazole) (rOPC), and
- 443 patients in other patient populations
These studies included immunocompromised patients (e.g., patients with hematological malignancy, neutropenia post-chemotherapy, GVHD post HSCT, and HIV infection), as well as non-neutropenic patients. This patient population was 71% male, had a mean age of 42 years (range: 8-84 years, 6% of patients were ≥65 years of age and 1% was <18 years of age), and were 64% White, 14% Black, and 16% Hispanic. Noxafil oral suspension therapy was given to 171 patients for ≥6 months, including 58 patients who received Noxafil oral suspension therapy for ≥12 months. Table 7 presents adverse reactions observed at an incidence of >10% in the studies for prophylaxis of invasive Aspergillus and Candida infections. Table 8 presents adverse reactions observed at an incidence of at least 10% in the OPC/rOPC studies.
Prophylaxis of Invasive Aspergillus and Candida Infections (Noxafil oral suspension)
In the two randomized, comparative studies for prophylaxis of invasive Aspergillus and Candida infections in those at high risk of developing these infections due to being severely immunocompromised (Noxafil Oral Suspension Study 1 and 2), the safety of Noxafil oral suspension 200 mg three times a day was compared to fluconazole 400 mg once daily or itraconazole 200 mg twice a day in severely immunocompromised patients. The most frequently reported adverse reactions (>30%) in these trials were fever, diarrhea, and nausea. The most common adverse reactions leading to discontinuation of Noxafil oral suspension were GI adverse reactions, specifically, nausea (2%), vomiting (2%), and hepatic enzymes increased (2%).
| Adverse Reactions | Noxafil Oral Suspension n=605 (%) | Fluconazole n=539 (%) | Itraconazole n=58 (%) |
|---|---|---|---|
| Percentage of Patients Reporting any Adverse Reaction | 98 | 99 | 100 |
| Fever | 45 | 47 | 55 |
| Diarrhea | 42 | 39 | 60 |
| Nausea | 38 | 37 | 52 |
| Hypokalemia | 30 | 26 | 52 |
| Thrombocytopenia | 29 | 27 | 34 |
| Vomiting | 29 | 32 | 41 |
| Headache | 28 | 26 | 40 |
| Abdominal Pain | 27 | 27 | 36 |
| Anemia | 25 | 23 | 28 |
| Coughing | 24 | 24 | 24 |
| Neutropenia | 23 | 23 | 40 |
| Constipation | 21 | 17 | 17 |
| Dyspnea | 20 | 22 | 26 |
| Rigors | 20 | 16 | 29 |
| Rash | 19 | 18 | 43 |
| Hypertension | 18 | 16 | 5 |
| Hypomagnesemia | 18 | 16 | 19 |
| Fatigue | 17 | 18 | 9 |
| Insomnia | 17 | 17 | 19 |
| Musculoskeletal Pain | 16 | 15 | 16 |
| Anorexia | 15 | 17 | 28 |
| Edema Legs | 15 | 12 | 19 |
| Epistaxis | 14 | 14 | 21 |
| Hypotension | 14 | 15 | 17 |
| Pharyngitis | 12 | 11 | 21 |
| Tachycardia | 12 | 14 | 5 |
| Arthralgia | 11 | 12 | 9 |
| Dizziness | 11 | 10 | 9 |
| Hyperglycemia | 11 | 14 | 3 |
| Petechiae | 11 | 10 | 16 |
| Pruritus | 11 | 12 | 19 |
| Back Pain | 10 | 12 | 7 |
| Bilirubinemia | 10 | 9 | 19 |
| Dyspepsia | 10 | 9 | 10 |
| Vaginal Hemorrhage Percentages of sex-specific adverse reactions are based on the number of males/females. | 10 | 9 | 12 |
Treatment of Nonrefractory OPC and Refractory OPC (Noxafil oral suspension)
In two randomized comparative studies for the treatment of nonrefractory OPC, the safety of Noxafil oral suspension (less than or equal to 400 mg once daily) in 557 HIV-infected patients was compared to the safety of fluconazole (100 mg once daily) in 262 HIV-infected patients.
An additional 239 HIV-infected patients with refractory OPC (rOPC) received Noxafil oral suspension in two non-comparative trials for rOPC. Of these patients, 149 received the 800 mg/day dosage and the remainder received the less than or equal to 400 mg once daily dosage.
In the nonrefractory OPC and rOPC studies, the most common adverse reactions in patients treated with Noxafil oral suspension were fever, diarrhea, nausea, headache, vomiting, and coughing.
Adverse reactions were reported more frequently in the studies of patients with refractory OPC. Among these highly immunocompromised patients with advanced HIV disease, serious adverse reactions were reported in 55% (132/239) of Noxafil oral suspension-treated patients. The most commonly reported serious adverse reactions were fever (13%) and neutropenia (10%).
| Adverse Reactions | Controlled OPC Pool | Refractory OPC Pool | |
|---|---|---|---|
| Noxafil Oral Suspension | Fluconazole | Noxafil Oral Suspension | |
| n=557 (%) | n=262 (%) | n=239 (%) | |
| OPC=oropharyngeal candidiasis | |||
| Percentage of Patients that Reported any Adverse Reaction Based on patients reporting adverse reactions at least once during the study, without regard to relationship to treatment. Patients may have reported more than 1 adverse reaction. | 64 | 67 | 92 |
| Diarrhea | 10 | 13 | 29 |
| Nausea | 9 | 11 | 29 |
| Headache | 8 | 9 | 20 |
| Vomiting | 7 | 7 | 28 |
| Fever | 6 | 8 | 34 |
| Abdominal Pain | 5 | 6 | 18 |
| Neutropenia | 4 | 3 | 16 |
| Coughing | 3 | 4 | 25 |
| Fatigue | 3 | 5 | 13 |
| Herpes Simplex | 3 | 3 | 11 |
| Pneumonia | 3 | 2 | 10 |
| Rash | 3 | 4 | 15 |
| Anemia | 2 | 2 | 14 |
| Anorexia | 2 | 2 | 19 |
| Asthenia | 2 | 2 | 13 |
| Sweating Increased | 2 | 2 | 10 |
| Candidiasis, Oral | 1 | <1 | 12 |
| Dehydration | 1 | 3 | 11 |
| Dyspnea | 1 | 3 | 12 |
| Insomnia | 1 | 1 | 16 |
| Pain | 1 | 1 | 11 |
| Weight Decrease | 1 | <1 | 14 |
| Rigors | <1 | 2 | 12 |
Additional Adverse Reactions Reported in Less Than 5% of Noxafil-Treated Patients in Clinical Trials
Other clinically significant adverse reactions reported in less than 5% of patients in clinical trials of Noxafil are listed below:
- Blood and lymphatic system disorders: hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, neutropenia aggravated
- Endocrine disorders: adrenal insufficiency
- Nervous system disorders: paresthesia
- Immune system disorders: allergic reaction [see Contraindications (4.1) ]
- Cardiac disorders: torsades de pointes [see Warnings and Precautions (5.2) ]
- Vascular disorders: pulmonary embolism
- Gastrointestinal disorders: pancreatitis
- Liver and Biliary System Disorders: hepatic enzymes increased, hepatic function abnormal, hepatitis, hepatomegaly, jaundice
- Renal & Urinary System Disorders: renal failure acute
Liver Test Abnormalities in the Clinical Trials of Noxafil Oral Suspension
Liver Test Abnormalities in the Clinical Trials with Noxafil Oral Suspension for Prophylaxis of Invasive Aspergillus and Candida Infections
In the prophylaxis of invasive Aspergillus and Candida infections studies, the number and percentage of patients with changes in liver tests from Common Toxicity Criteria (CTC) Grade 0, 1, or 2 at baseline to Grade 3 or 4 at the end of the studies is presented in Table 9 .
| Number (%) of Patients with Change Change from Grade 0 to 2 at baseline to Grade 3 or 4 during the study. These data are presented in the form X/Y, where X represents the number of patients who met the criterion as indicated, and Y represents the number of patients who had a baseline observation and at least one post-baseline observation. | ||
|---|---|---|
| CTC = Common Toxicity Criteria; AST= Aspartate Aminotransferase; ALT= Alanine Aminotransferase. | ||
| Noxafil Oral Suspension Study 1 | ||
| Laboratory Parameter | Noxafil Oral Suspension n=301 | Fluconazole n=299 |
| AST | 11/266 (4) | 13/266 (5) |
| ALT | 47/271 (17) | 39/272 (14) |
| Bilirubin | 24/271 (9) | 20/275 (7) |
| Alkaline Phosphatase | 9/271 (3) | 8/271 (3) |
| Noxafil Oral Suspension Study 2 | ||
| Laboratory Parameter | Noxafil Oral Suspension (n=304) | Fluconazole/Itraconazole (n=298) |
| AST | 9/286 (3) | 5/280 (2) |
| ALT | 18/289 (6) | 13/284 (5) |
| Bilirubin | 20/290 (7) | 25/285 (9) |
| Alkaline Phosphatase | 4/281 (1) | 1/276 (<1) |
Liver Test Abnormalities in the Clinical Trials with Noxafil Oral Suspension for the Treatment of OPC
The number and percentage of patients treated for OPC with clinically significant liver test abnormalities at any time during the studies is provided in Table 10 (liver test abnormalities were present in some of these patients prior to initiation of the study drug).
| Laboratory Test | Nonrefractory OPC | Refractory OPC | |
|---|---|---|---|
| Noxafil Oral Suspension | Fluconazole | Noxafil Oral Suspension | |
| n=557 (%) | n=262 (%) | n=239 (%) | |
| ALT= Alanine Aminotransferase; AST= Aspartate Aminotransferase. | |||
| ALT > 3.0 x ULN | 16/537 (3) | 13/254 (5) | 25/226 (11) |
| AST > 3.0 x ULN | 33/537 (6) | 26/254 (10) | 39/223 (17) |
| Total Bilirubin > 1.5 x ULN | 15/536 (3) | 5/254 (2) | 9/197 (5) |
| Alkaline Phosphatase > 3.0 x ULN | 17/535 (3) | 15/253 (6) | 24/190 (13) |
Liver Test Abnormalities in the Clinical Trials with Noxafil Oral Suspension for the Treatment of Invasive Aspergillosis
The number and percentage of patients treated for invasive aspergillosis with clinically significant liver test abnormalities at any time during the Aspergillosis Treatment Study is provided in Table 11. Liver test abnormalities present prior to the initiation of study drug included ALT (22% of the patients), AST (13% of the patients), and bilirubin (13% of the patients).
| Number (%) of Patients with Change Change from Grade 0 to 2 at baseline to Grade 3 or 4 during the study. These data are presented in the form n/N, where n represents the number of patients who met the criterion as indicated, and N represents the number of patients who had a baseline observation and at least one post-baseline observation. | ||
|---|---|---|
| Laboratory Parameter | Noxafil n/N (%) | Voriconazole n/N (%) |
| N=Number of patients for a given laboratory test with a baseline value of CTC Grade 0, 1, or 2 and at least one post-baseline value. CTC = Common Toxicity Criteria; AST= Aspartate Aminotransferase; ALT= Alanine Aminotransferase. | ||
| AST | 22/281 (8) | 21/285 (7) |
| ALT | 29/281(10) | 23/282 (8) |
| Bilirubin | 26/280 (9) | 25/284 (9) |
| Alkaline Phosphatase | 12/282 (4) | 20/284 (7) |
In healthy volunteers and patients, elevation of liver test values did not appear to be associated with higher plasma concentrations of posaconazole.
Clinical Trials in Pediatric Patients 2 Years of Age and Older
The safety of Noxafil injection and Noxafil PowderMix (for delayed-release oral suspension) for prophylaxis of invasive fungal infections was evaluated in an open-label uncontrolled dose-ranging pharmacokinetic and safety study of Noxafil injection and Noxafil PowderMix (Pediatric Study 1, NCT02452034). In this study, 115 immunocompromised pediatric patients 2 to less than 18 years of age with known or expected neutropenia initially received Noxafil injection (up to 6 mg/kg twice daily for the first day and then up to 6 mg/kg for at least 7 days) and then 63 patients were transitioned to Noxafil PowderMix (up to 6 mg/kg once daily). The mean overall treatment duration for all treated subjects was 21 days including a mean duration of 14 days (range: 1 to 28 days) on Noxafil injection and a mean duration of 12 days (range: 2 to 18 days) on Noxafil PowderMix for delayed-release oral suspension . In this study, the reported adverse reaction profile of Noxafil injection and Noxafil PowderMix in pediatric patients was consistent with the safety profile of Noxafil in adults.
The safety of Noxafil injection, Noxafil delayed-release tablets, and Noxafil PowderMix for delayed-release oral suspension for the treatment of invasive aspergillosis was evaluated in an open-label, non-comparative clinical study in 31 pediatric patients 2 to less than 18 years of age with a diagnosis of possible, probable, or proven invasive aspergillosis (Pediatric Study 2, NCT04218851). In this study, all 31 pediatric patients initially received Noxafil injection (6 mg/kg twice daily on the first day and then 6 mg/kg once daily) for the treatment of invasive aspergillosis; 12 patients were transitioned to Noxafil delayed-release tablets (300 mg once daily) if they weighed ≥40 kg, and 10 patients were transitioned to Noxafil PowderMix (based on weight) if they weighed 10 to 40 kg [see Dosage and Administration (2.3) ] . The mean overall treatment duration was 50 days including 15 days (range: 2 to 78 days) on Noxafil injection, 54 days (range: 6 to 80 days) on Noxafil delayed-release tablets, and 44 days (range: 7 to 76 days) on Noxafil PowderMix. The reported adverse reaction profile of Noxafil injection, Noxafil delayed-release tablets, and Noxafil PowderMix in pediatric patients was consistent with the known safety profile of Noxafil in adults. The most common adverse reactions that occurred in greater than 20% of pediatric patients who received any of the three formulations of Noxafil were vomiting, pyrexia, abdominal pain, liver test abnormalities, and hypertension.
Postmarketing Experience
The following adverse reaction has been identified during the post-approval use of posaconazole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a casual relationship to drug exposure.
Endocrine Disorders: Pseudoaldosteronism
DRUG INTERACTIONS
Table 12 and Table 13 include drugs with clinically important drug interactions when administered concomitantly with posaconazole and instructions for preventing or managing them. Table 14 includes important drug interactions specific to the absorption of posaconazole administered as Posaconazole oral suspension.
These recommendations are based on either drug interaction studies or predicted interactions due to the expected magnitude of interaction and potential for serious adverse reactions or loss of efficacy [see Clinical Pharmacology (12.3) ] .
The following information was derived from data with Noxafil oral suspension or another posaconazole tablet formulation unless otherwise noted. All clinically important drug interactions with Posaconazole oral suspension, except for those that affect the absorption of posaconazole (via gastric pH and motility), are considered relevant to clinically important drug interactions with Posaconazole delayed-release tablets [see Clinical Pharmacology (12.3) ] .
Consult the labeling of concomitantly used drugs to obtain further information about interactions with posaconazole.
Effects of Other Drugs on Posaconazole
Posaconazole is primarily metabolized via UDP-glucuronosyltransferase and is a substrate of p-glycoprotein (P-gp) efflux. Therefore, inhibitors or inducers of these clearance pathways may affect posaconazole plasma concentrations. Concomitant use of posaconazole with drugs that can decrease the plasma posaconazole concentrations should generally be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
| UDP-Glucuronidase Inducers | ||
|---|---|---|
| Mechanism and Clinical Effect(s) | Posaconazole is a UDP-glucuronosyltransferase substrate. Concomitant use of posaconazole with UDP-glucuronidase inducers may decrease posaconazole exposure [see Clinical Pharmacology (12.3) ], which may reduce the effectiveness of posaconazole. | |
| Prevention or Management | Efavirenz | Avoid concomitant use of posaconazole with efavirenz, unless the benefit outweighs the risks. |
| Rifabutin | Avoid concomitant use of posaconazole with rifabutin unless the benefit to the patient outweighs the risk. If concomitant use is needed, monitor closely for breakthrough fungal infections. See Table 17 for rifabutin monitoring considerations when posaconazole affects rifabutin via CYP3A4 inhibition. | |
| Phenytoin | Avoid concomitant use of posaconazole with phenytoin unless the benefit to the patient outweighs the risk. If concomitant use is needed, monitor for breakthrough fungal infections. See Table 17 for phenytoin monitoring considerations when posaconazole affects phenytoin via CYP3A4 inhibition. | |
| Fosamprenavir | ||
| Mechanism and Clinical Effect(s) | Concomitant use of posaconazole with fosamprenavir may lead to decreased posaconazole plasma concentrations [see Clinical Pharmacology (12.3) ], which may reduce effectiveness of posaconazole. | |
| Prevention or Management | If concomitant use of posaconazole with fosamprenavir is needed, monitor closely for breakthrough fungal infections. | |
| Posaconazole Oral Suspension | |
|---|---|
| Cimetidine and Esomeprazole | |
| Mechanism and Clinical Effect(s) | Concomitant use of Noxafil oral suspension with cimetidine or esomeprazole resulted in decreased posaconazole plasma concentrations [see Clinical Pharmacology (12.3) ], which may reduce effectiveness of posaconazole. |
| Prevention or Management | Avoid concomitant use of posaconazole oral suspension with cimetidine or esomeprazole unless the benefit outweighs the risks. If concomitant use is needed, monitor closely for breakthrough fungal infections. |
| Metoclopramide | |
| Mechanism and Clinical Effect(s) | Concomitant use of Noxafil oral suspension with metoclopramide decreased posaconazole plasma concentrations [see Clinical Pharmacology (12.3) ], which may reduce effectiveness of Posaconazole oral suspension. |
| Prevention or Management | If Posaconazole oral suspension is concomitantly administered with metoclopramide, closely monitor for breakthrough fungal infections. |
Effects of Posaconazole on Other Drugs
Posaconazole is a strong CYP3A4 inhibitor. Therefore, concomitant use of posaconazole may increase plasma concentrations of drugs that are CYP3A4 substrates [see Clinical Pharmacology (12.3) ] .
| Digoxin | ||
|---|---|---|
| Clinical Effect(s) | Increased digoxin plasma concentrations have been reported in patients who received concomitant posaconazole and digoxin. | |
| Prevention or Management | Monitor digoxin plasma concentrations during concomitant use of posaconazole. | |
| Glipizide | ||
| Clinical Effect(s) | No dosage modification of glipizide is needed when used concomitantly with posaconazole. However, glucose concentrations decrease in some patients concomitantly administered posaconazole and glipizide. | |
| Prevention or Management | Increase monitoring of glucose concentrations when used concomitantly. | |
| CYP3A Substrates | ||
| Immunosuppressants that are CYP3A4 Substrates | ||
| Mechanism and Clinical Effect(s) | Posaconazole is a strong CYP3A4 inhibitor. Therefore, plasma concentrations of CYP3A4 substrates may be increased by posaconazole use [see Clinical Pharmacology (12.3) ]. | |
| Prevention or Management | Sirolimus | Posaconazole is contraindicated with sirolimus [see Clinical Pharmacology (12.3) ] . |
| Tacrolimus |
| |
| Cyclosporine |
| |
| CYP3A4 Substrates that Prolong QTc Interval | ||
| Mechanism and Clinical Effect(s) | Concomitant use of posaconazole with CYP3A4 substrates such as pimozide and quinidine may result in increased plasma concentrations of the CYP3A4 substrates leading to QTc interval prolongation and torsades de pointes [see Warnings and Precautions (5.2) ]. | |
| Prevention or Management | Pimozide | Concomitant use with posaconazole is contraindicated. |
| Quinidine | ||
| HMG-CoA Reductase Inhibitors (Statins) that are CYP3A4 Substrates | ||
| Mechanism and Clinical Effect(s) | Concomitant use of posaconazole with simvastatin increased simvastatin plasma concentrations which can lead to rhabdomyolysis [see Clinical Pharmacology (12.3) ]. | |
| Prevention or Management | Atorvastatin, Lovastatin, Simvastatin | Concomitant use with posaconazole is contraindicated. |
| Benzodiazepines that are CYP3A4 Substrates | ||
| Mechanism and Clinical Effect(s) | Concomitant use of posaconazole with midazolam increased midazolam plasma concentrations which could potentiate and prolong hypnotic and sedative effects [see Clinical Pharmacology (12.3) ] . | |
| Prevention or Management | Midazolam, Alprazolam, Triazolam | Closely monitor for adverse reactions associated with high plasma concentrations of benzodiazepines that are CYP3A4 substrates during concomitant use, and a benzodiazepine receptor antagonist should be available to reverse effects [see Warnings and Precautions (5.7) ]. |
| Calcium Channel Blockers that are CYP3A4 Substrates | ||
| Mechanism and Clinical Effect(s) | Posaconazole may increase the plasma concentrations of calcium channel blockers that are substrates of CYP3A4. | |
| Prevention or Management | Verapamil, Diltiazem, Nifedipine, Nicardipine, Felodipine | Monitor frequently for adverse reactions and toxicity with concomitant use of posaconazole with calcium channel blockers that are CYP3A4 substrates. Dosage reduction of the calcium channel blocker may be needed. |
| Anti-HIV Drugs that are CYP3A4 Substrates | ||
| Mechanism and Clinical Effect(s) | Ritonavir and atazanavir are CYP3A4 substrates and posaconazole increased plasma concentrations of these drugs [see Clinical Pharmacology (12.3) ]. | |
| Prevention or Management | Ritonavir and Atazanavir | Monitor frequently for adverse reactions and toxicity of ritonavir and atazanavir during concomitant use. |
| Antineoplastic Drugs that are CYP3A4 Substrates | ||
| Mechanism and Clinical Effect(s) | Posaconazole may increase plasma concentrations of oncology drugs that are CYP3A4 substrates, which may increase the risk of serious adverse reactions. | |
| Prevention or Management | Venetoclax | CLL/SLL patients: Concomitant use of posaconazole with venetoclax during initiation and ramp-up phase is contraindicated. AML patients: With concomitant use, venetoclax dosage reduction and safety monitoring is recommended across all dosing phases [see Warnings and Precautions (5.11) ] . |
| Vinca alkaloids (e.g., vincristine, vinblastine) | Reserve concomitant use for patients with no alternative antifungal treatment options [see Warnings and Precautions (5.8) ]. | |
| Ergot Alkaloids | ||
| Mechanism and Clinical Effect(s) | Most of the ergot alkaloids are CYP3A4 substrates. Posaconazole may increase the plasma concentrations of ergot alkaloids (ergotamine and dihydroergotamine) which may lead to ergotism. | |
| Prevention or Management | Ergotamine, Dihydroergotamine | Concomitant use with posaconazole is contraindicated. |
| Phenytoin | ||
| Mechanism and Clinical Effect(s) | Phenytoin is a CYP3A4 substrate. Concomitant use of posaconazole with phenytoin increased phenytoin plasma concentrations [see Clinical Pharmacology (12.3) ]. | |
| Prevention or Management | Avoid concomitant use of posaconazole with phenytoin unless the benefit outweighs the risk. frequently monitor phenytoin concentrations and consider a dosage reduction of phenytoin. See Table 15 for additional monitoring considerations when phenytoin affects posaconazole via UDP-glucuronosyltransferase inhibition. | |
| Rifabutin | ||
| Mechanism and Clinical Effect(s) | Rifabutin is a CYP3A4 substrate. Concomitant use of posaconazole with rifabutin increased rifabutin plasma concentrations [see Clinical Pharmacology (12.3) ]. | |
| Prevention or Management | Avoid concomitant use of posaconazole with rifabutin unless the benefit outweighs the risk. Frequent monitoring of full blood counts and adverse reactions due to increased rifabutin plasma concentrations (e.g., uveitis, leukopenia) during concomitant use are recommended. See Table 15 for additional monitoring considerations when rifabutin affects posaconazole via UDP-glucuronosyltransferase inhibition. | |
Absence of Clinically Important Interaction with Posaconazole
Additional clinical studies demonstrated that no clinically important effects on zidovudine, lamivudine, indinavir, or caffeine were observed when administered with Noxafil 200 mg once daily; therefore, no dose adjustments are required for these drugs when coadministered with posaconazole 200 mg once daily.
No clinically relevant effects on the pharmacokinetics of Noxafil delayed-release tablets were observed during concomitant use with antacids, H 2 -receptor antagonists and proton pump inhibitors, and metoclopramide [see Clinical Pharmacology (12.3) ] . No dosage adjustment of Posaconazole delayed-release tablets is required during concomitant use with these drugs.
No clinically relevant effects on the pharmacokinetics of Noxafil oral suspension were observed during concomitant use with antacids, H 2 -receptor antagonists (other than cimetidine), and loperamide [see Clinical Pharmacology (12.3) ] . No dosage adjustment of Posaconazole oral suspension is required during concomitant use with these drugs (other than cimetidine).
DESCRIPTION
Posaconazole delayed-release tablets and Posaconazole oral suspension contain posaconazole; an azole antifungal agent.
Posaconazole is designated chemically as 4-[4-[4-[4-[[ (3 R ,5 R )-5- (2,4-difluorophenyl)tetrahydro-5- (1 H -1,2,4-triazol-1-ylmethyl)-3-furanyl]methoxy]phenyl]-1-piperazinyl]phenyl]-2-[(1 S ,2 S )-1-ethyl-2-hydroxypropyl]-2,4-dihydro-3 H -1,2,4-triazol-3-one with an empirical formula of C 37 H 42 F 2 N 8 O 4 and a molecular weight of 700.8. The chemical structure is:
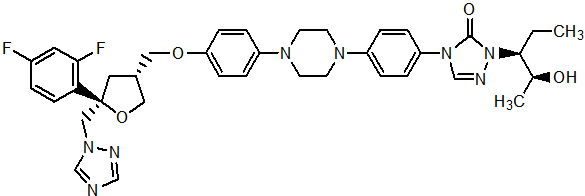
Posaconazole is a white powder with a low aqueous solubility.
Posaconazole Delayed-Release Tablets
Posaconazole delayed-release tablet, for oral use, is yellow, coated, and oblong and contains 100 mg of posaconazole. Each delayed-release tablet contains the inactive ingredients: croscarmellose sodium, hydroxypropylcellulose, hypromellose acetate succinate, iron oxide yellow, Macrogol/PEG 3350, magnesium stearate, microcrystalline cellulose, polyvinyl alcohol partially hydrolyzed, silicon dioxide, talc, and titanium dioxide.
Posaconazole Oral Suspension
Posaconazole oral suspension is a white, cherry-flavored immediate-release suspension that contains 40 mg of posaconazole per mL and the following inactive ingredients: artificial cherry flavor, citric acid monohydrate, glycerin, liquid glucose, polysorbate 80, purified water, simethicone, sodium benzoate, sodium citrate dihydrate, titanium dioxide, and xanthan gum.
CLINICAL PHARMACOLOGY
Mechanism of Action
Posaconazole is an azole antifungal agent [see Clinical Pharmacology (12.4) ] .
Pharmacodynamics
Exposure Response Relationship: Prophylaxis of invasive Aspergillus and Candida Infections in Adults
Who Are at High Risk of Developing These Infections Due to Being Severely Immunocompromised
In clinical studies of neutropenic patients who were receiving cytotoxic chemotherapy for acute myelogenous leukemia (AML) or myelodysplastic syndromes (MDS) or hematopoietic stem cell transplant (HSCT) recipients with Graft versus Host Disease (GVHD), a wide range of plasma posaconazole exposures was noted following administration of Noxafil oral suspension. A pharmacokinetic-pharmacodynamic analysis of patient data revealed an apparent association between average posaconazole concentrations (Cavg) and prophylactic efficacy (Table 15). A lower Cavg may be associated with an increased risk of treatment failure, defined as treatment discontinuation, use of empiric systemic antifungal therapy (SAF), or occurrence of breakthrough invasive fungal infections.
| Prophylaxis in AML/MDS Neutropenic patients who were receiving cytotoxic chemotherapy for AML or MDS | Prophylaxis in GVHD HSCT recipients with GVHD | |||
|---|---|---|---|---|
| Cavg Range (ng/mL) | Treatment Failure Defined as treatment discontinuation, use of empiric systemic antifungal therapy (SAF), or occurrence of breakthrough invasive fungal infections (%) | Cavg Range (ng/mL) | Treatment Failure(%) | |
| Cavg = the average posaconazole concentration when measured at steady state | ||||
| Quartile 1 | 90-322 | 54.7 | 22-557 | 44.4 |
| Quartile 2 | 322-490 | 37.0 | 557-915 | 20.6 |
| Quartile 3 | 490-734 | 46.8 | 915-1563 | 17.5 |
| Quartile 4 | 734-2200 | 27.8 | 1563-3650 | 17.5 |
Exposure Response Relationship: Treatment of Invasive Aspergillosis in Adult and Adolescent Patients:
Across a range of posaconazole plasma minimum concentrations (C min, range: 244 to 5663 ng/mL) following administration of Noxafil injection and Noxafil delayed-release tablets in adult and pediatric patients aged 14 years and older treated for invasive aspergillosis in Aspergillosis Treatment Study, there was no association between posaconazole C min and treatment efficacy [see Clinical Pharmacology (12.3) and Clinical Studies (14.1) ] . Similarly, across a range of population pharmacokinetic model-predicted steady-state plasma average concentrations (Cavg, range: 589 to 6315 ng/mL), there was no association between posaconazole Cavg and treatment efficacy.
Pharmacokinetics
General Pharmacokinetic Characteristics
General Pharmacokinetic Characteristics of Posaconazole Delayed-Release Tablets
Noxafil delayed-release tablets exhibit dose proportional pharmacokinetics after single and multiple dosing up to 300 mg. The mean pharmacokinetic parameters of posaconazole at steady state following administration of Noxafil delayed-release tablets 300 mg twice daily on Day 1, then 300 mg once daily thereafter in healthy volunteers and in neutropenic patients who are receiving cytotoxic chemotherapy for AML or MDS or HSCT recipients with GVHD are shown in Table 16.
| N | AUC 0-24 hr (ng∙hr/mL) | Cav Cav = time-averaged concentrations (i.e., AUC 0-24 hr /24 hr) (ng/mL) | C max (ng/mL) | C min (ng/mL) | T max Median (minimum-maximum) (hr) | t 1/2 (hr) | CL/F (L/hr) | |
|---|---|---|---|---|---|---|---|---|
| CV = coefficient of variation expressed as a percentage (%CV); AUC 0-T = Area under the plasma concentration-time curve from time zero to 24 hr; C max = maximum observed concentration; C min = minimum observed plasma concentration; T max = time of maximum observed concentration; t ½ = terminal phase half-life; CL/F = Apparent total body clearance | ||||||||
| Healthy Volunteers | 12 | 51618 (25) | 2151 (25) | 2764 (21) | 1785 (29) | 4 (3-6) | 31 (40) | 7.5 (26) |
| Patients | 50 | 37900 (42) | 1580 (42) | 2090 (38) | 1310 (50) | 4 (1.3-8.3) | - | 9.39 (45) |
General Pharmacokinetic Characteristics of Posaconazole Oral Suspension
Dose-proportional increases in plasma exposure (AUC) to Noxafil oral suspension were observed following single oral doses from 50 mg to 800 mg and following multiple-dose administration from 50 mg twice daily to 400 mg twice daily in healthy volunteers. No further increases in exposure were observed when the dose of the oral suspension increased from 400 mg twice daily to 600 mg twice daily in febrile neutropenic patients or those with refractory invasive fungal infections.
The mean (%CV) [min-max] Noxafil oral suspension average steady-state plasma concentrations (Cavg) and steady-state pharmacokinetic parameters in patients following administration of 200 mg three times a day and 400 mg twice daily of the oral suspension are provided in Table 17.
| Dose Oral suspension administration | Cavg (ng/mL) | AUC AUC (0-24 hr) for 200 mg three times a day and AUC (0-12 hr) for 400 mg twice daily (ng∙hr/mL) | CL/F (L/hr) | V/F (L) | t ½ (hr) |
|---|---|---|---|---|---|
| Cavg = the average posaconazole concentration when measured at steady state | |||||
| 200 mg three times a day HSCT recipients with GVHD (n=252) | 1103 (67) [21.5-3650] | ND Not done | ND | ND | ND |
| 200 mg three times a day Neutropenic patients who were receiving cytotoxic chemotherapy for acute myelogenous leukemia or myelodysplastic syndromes (n=215) | 583 (65) [89.7-2200] | 15,900 (62) [4100-56,100] | 51.2 (54) [10.7-146] | 2425 (39) [828-5702] | 37.2 (39) [19.1-148] |
| 400 mg twice daily Febrile neutropenic patients or patients with refractory invasive fungal infections, Cavg n=24 The variability in average plasma posaconazole concentrations in patients was relatively higher than that in healthy subjects. (n=23) | 723 (86) [6.70-2256] | 9093 (80) [1564-26,794] | 76.1 (78) [14.9-256] | 3088 (84) [407-13,140] | 31.7 (42) [12.4-67.3] |
Absorption:
Absorption of Posaconazole Delayed-Release Tablets
When given orally in healthy volunteers, Noxafil delayed-release tablets are absorbed with a median T max of 4 to 5 hours. Steady-state plasma concentrations are attained by Day 6 at the 300 mg dose (once daily after twice daily loading dose at Day 1). The absolute bioavailability of the oral delayed-release tablet is approximately 54% under fasted conditions. The C max and AUC of posaconazole following administration of Noxafil delayed-release tablets are increased 16% and 51%, respectively, when given with a high fat meal compared to a fasted state (see Table 18 ).
| Fasting Conditions | Fed Conditions (High Fat Meal) 48.5 g fat | Fed/Fasting | |||
|---|---|---|---|---|---|
| Pharmacokinetic Parameter | N | Mean (%CV) | N | Mean (%CV) | GMR (90% CI) |
| GMR=Geometric least-squares mean ratio; CI=Confidence interval | |||||
| C max (ng/mL) | 14 | 935 (34) | 16 | 1060 (25) | 1.16 (0.96, 1.41) |
| AUC 0-72hr (hr∙ng/mL) | 14 | 26200 (28) | 16 | 38400 (18) | 1.51 (1.33, 1.72) |
| T max Median (Min, Max) reported for T max (hr) | 14 | 5.00 (3.00, 8.00) | 16 | 6.00 (5.00, 24.00) | N/A |
Absorption of Posaconazole Oral Suspension
Noxafil oral suspension is absorbed with a median T max of ~3 to 5 hours. Steady-state plasma concentrations are attained at 7 to 10 days following multiple-dose administration.
Following single-dose administration of 200 mg, the mean AUC and C max of posaconazole are approximately 3-times higher when the oral suspension is administered with a nonfat meal and approximately 4-times higher when administered with a high-fat meal (~50 gm fat) relative to the fasted state. Following single-dose administration of Noxafil oral suspension 400 mg, the mean AUC and C max of posaconazole are approximately 3-times higher when administered with a liquid nutritional supplement (14 gm fat) relative to the fasted state (see Table 19 ). In addition, the effects of varying gastric administration conditions on the C max and AUC of Noxafil oral suspension in healthy volunteers have been investigated and are shown in Table 20.
To assure attainment of adequate plasma concentrations, it is recommended to administer Posaconazole oral suspension during or immediately following a full meal. In patients who cannot eat a full meal, Posaconazole oral suspension should be taken with a liquid nutritional supplement or an acidic carbonated beverage (e.g., ginger ale).
| Dose (mg) | C max (ng/mL) | T max Median [min-max]. (hr) | AUC (I) (ng∙hr/mL) | CL/F (L/hr) | t ½ (hr) |
|---|---|---|---|---|---|
| 200 mg fasted (n=20) n=15 for AUC (I), CL/F, and t ½ | 132 (50) [45-267] | 3.50 [1.5-36 The subject with T max of 36 hrs had relatively constant plasma levels over 36 hrs (1.7 ng/mL difference between 4 hrs and 36 hrs). ] | 4179 (31) [2705-7269] | 51 (25) [28-74] | 23.5 (25) [15.3-33.7] |
| 200 mg nonfat (n=20) | 378 (43) [131-834] | 4 [3-5] | 10,753 (35) [4579-17,092] | 21 (39) [12-44] | 22.2 (18) [17.4-28.7] |
| 200 mg high fat (54 gm fat) (n=20) | 512 (34) [241-1016] | 5 [4-5] | 15,059 (26) [10,341-24,476] | 14 (24) [8.2-19] | 23.0 (19) [17.2-33.4] |
| 400 mg fasted (n=23) n=10 for AUC (I), CL/F, and t ½ | 121 (75) [27-366] | 4 [2-12] | 5258 (48) [2834-9567] | 91 (40) [42-141] | 27.3 (26) [16.8-38.9] |
| 400 mg with liquid nutritional supplement (14 gm fat) (n=23) | 355 (43) [145-720] | 5 [4-8] | 11,295 (40) [3865-20,592] | 43 (56) [19-103] | 26.0 (19) [18.2-35.0] |
| Study Description | Administration Arms | Change in C max (ratio estimate Ratio Estimate is the ratio of coadministered drug plus Noxafil to coadministered drug alone for C max or AUC. ; 90% CI of the ratio estimate) | Change in AUC (ratio estimate; 90% CI of the ratio estimate) |
|---|---|---|---|
| 400-mg single dose with a high-fat meal relative to fasted state (n=12) | 5 minutes before high-fat meal | ↑96% (1.96; 1.48-2.59) | ↑111% (2.11; 1.60-2.78) |
| During high-fat meal | ↑339% (4.39; 3.32-5.80) | ↑382% (4.82; 3.66-6.35) | |
| 20 minutes after high-fat meal | ↑333% (4.33; 3.28-5.73) | ↑387% (4.87; 3.70-6.42) | |
| 400 mg twice daily and 200 mg four times daily for 7 days in fasted state and with liquid nutritional supplement (BOOST ® ) (n=12) | 400 mg twice daily with BOOST | ↑65% (1.65; 1.29-2.11) | ↑66% (1.66; 1.30-2.13) |
| 200 mg four times daily with BOOST | No Effect | No Effect | |
| Divided daily dose from 400 mg twice daily to 200 mg four times daily for 7 days regardless of fasted conditions or with BOOST (n=12) | Fasted state | ↑136% (2.36; 1.84-3.02) | ↑161% (2.61; 2.04-3.35) |
| With BOOST | ↑137% (2.37; 1.86-3.04) | ↑157% (2.57; 2.00-3.30) | |
| 400-mg single dose with carbonated acidic beverage (ginger ale) and/or proton pump inhibitor (esomeprazole) (n=12) | Ginger ale | ↑92% (1.92; 1.51-2.44) | ↑70% (1.70; 1.43-2.03) |
| Esomeprazole | ↓32% (0.68; 0.53-0.86) | ↓30% (0.70; 0.59-0.83) | |
| 400-mg single dose with a prokinetic agent (metoclopramide 10 mg three times a day for 2 days) + BOOST or an antikinetic agent (loperamide 4-mg single dose) + BOOST (n=12) | With metoclopramide + BOOST | ↓21% (0.79; 0.72-0.87) | ↓19% (0.81; 0.72-0.91) |
| With loperamide + BOOST | ↓3% (0.97; 0.88-1.07) | ↑11% (1.11; 0.99-1.25) | |
| 400-mg single dose either orally with BOOST or via an NG tube with BOOST (n=16) | Via NG tube NG = nasogastric | ↓19% (0.81; 0.71-0.91) | ↓23% (0.77; 0.69-0.86) |
Distribution:
Posaconazole is highly bound to human plasma proteins (>98%), predominantly to albumin.
Metabolism:
Posaconazole primarily circulates as the parent compound in plasma. Of the circulating metabolites, the majority are glucuronide conjugates formed via UDP glucuronidation (phase 2 enzymes). Posaconazole does not have any major circulating oxidative (CYP450 mediated) metabolites. The excreted metabolites in urine and feces account for ~17% of the administered radiolabeled dose.
Posaconazole is a substrate for p-glycoprotein (P-gp) efflux.
In vitro studies with human hepatic microsomes and clinical studies indicate that posaconazole is an inhibitor primarily of CYP3A4.
Excretion:
Following administration of Noxafil oral suspension, posaconazole is predominantly eliminated in the feces (71% of the radiolabeled dose up to 120 hours) with the major component eliminated as parent drug (66% of the radiolabeled dose). Renal clearance is a minor elimination pathway, with 13% of the radiolabeled dose excreted in urine up to 120 hours (<0.2% of the radiolabeled dose is parent drug).
Posaconazole delayed-release tablet is eliminated with a mean half-life (t ½ ) ranging between 26 to 31 hours.
Posaconazole oral suspension is eliminated with a mean half-life (t ½ ) of 35 hours (range: 20-66 hours).
Specific Populations
No clinically significant differences in the pharmacokinetics of posaconazole were observed based on age, sex, renal impairment, and indication (prophylaxis or treatment).
Patients with Renal Impairment:
After Noxafil oral administration, there were no significant differences in the posaconazole pharmacokinetics in patients with eGFR 20 mL/minute/1.73 m 2 or higher compared to those with eGFR >80 mL/minute/1.73 m 2 . Although the mean posaconazole plasma exposure (AUC) was similar in patients with eGFR less than 20 mL/minute/1.73 m 2 treated with Noxafil oral suspension to those with eGFR >80 mL/minute/1.73 m 2 treated with Noxafil oral suspension, the range of the AUC estimates was highly variable (CV=96%) in patients with eGFR less than 20 mL/minute/1.73 m 2 compared to those with eGFR >80 mL/minute/1.73 m 2 (CV <40%).Similar posaconazole pharmacokinetic results are expected after administration of Posaconazole delayed-release tablets [see Use in Specific Populations (8.6) ].
Patients with Hepatic Impairment:
After a single oral dose of Noxafil oral suspension 400 mg, the mean AUC was 43%, 27%, and 21% higher in subjects with mild (Child-Pugh Class A, N=6), moderate (Child-Pugh Class B, N=6), or severe (Child-Pugh Class C, N=6) hepatic impairment, respectively, compared to subjects with normal hepatic function (N=18). Compared to subjects with normal hepatic function, the mean C max was 1% higher, 40% higher, and 34% lower in subjects with mild, moderate, or severe hepatic impairment, respectively [see Use in Specific Populations (8.7) ].
Race/Ethnicity:
In a population pharmacokinetic analysis of posaconazole, AUC was found to be 25% higher in Chinese patients relative to patients from other races/ethnicities. This higher exposure is not expected to be clinically relevant given the expected variability in posaconazole exposure [see Use in Specific Populations (8.9) ] .
Patients Weighing More Than 120 kg:
Weight has a clinically significant effect on posaconazole clearance. Relative to 70 kg patients, the Cavg is decreased by 25% in patients greater than 120 kg. Patients administered posaconazole weighing more than 120 kg may be at higher risk for lower posaconazole plasma concentrations compared to lower weight patients [see Use in Specific Populations (8.10) ] .
Pediatric Patients:
Treatment of invasive aspergillosis in pediatric patients 2 years of age and older: A total of 31 patients 2 to less than 18 years of age (body weight of ≥12 kg) received pediatric dosing based on body weight of Noxafil delayed-release tablets, Noxafil Injection, and Noxafil PowderMix for delayed-release oral suspension [see Dosage and Administration (2.3 ].
The mean population pharmacokinetic model parameters after multiple dose administration of Noxafil delayed-release tablets, Noxafil Injection, and Noxafil PowderMix for delayed-release oral suspension in pediatric patients 2 to less than 18 years of age for the treatment of invasive aspergillosis (Pediatric Study 2) are shown in Table 21. [see Adverse Reactions (6.1) ] .
| Age Group | Dose Type | N Some patients had 2 values (1 for IV dosing and 1 for oral dosing) | AUC 0-24 hours (ng·hr/mL) | C av Cav = time-averaged concentrations (i.e., AUC 0-24 hours /24hr) (ng/mL) | C max (ng/mL) | C min (ng/mL) | T max Median (minimum-maximum) (hr) | CL/F Clearance (CL for IV and CL/F for PFS or Tablet) (L/hr) |
|---|---|---|---|---|---|---|---|---|
| IV = Noxafil injection; PFS = Noxafil PowderMix for delayed-release oral suspension; Tablet= Noxafil delayed-release tablets; AUC0-24 hours = Area under the plasma concentration-time curve from time zero to 24 hr; C max = maximum observed concentration; C min = minimum observed plasma concentration; T max = time of maximum observed concentration; CL/F = apparent total body clearance | ||||||||
| 2 to <12 years | IV | 9 | 61900 (49.8) | 2580 (49.8) | 3630 (30.8) | 1710 (82.2) | 1.50 (1.25-1.77) | 2.56 (47.8) |
| PFS | 6 | 45200 (30.2) | 1880 (30.2) | 2220 (26.5) | 1370 (41.8) | 7.00 (6.40-7.20) | 3.25 (34.6) | |
| 12 to <18 years | IV | 13 | 60800 (35.6) | 2530 (35.6) | 3510 (26.8) | 1740 (48.5) | 1.50 (1.30-1.63) | 4.41 (41.8) |
| Tablet | 10 | 47800 (52.7) | 1990 (52.7) | 2250 (48.3) | 1580 (62.6) | 7.15 (6.70-7.30) | 6.27 (52.7) | |
The population pharmacokinetic analysis of posaconazole in pediatric patients, including Pediatric Study 2, suggests that age, sex, ethnicity, and disease status have no clinically meaningful effect on the pharmacokinetics of posaconazole.
Drug Interaction Studies:
Posaconazole is primarily metabolized via UDP glucuronidation (phase 2 enzymes) and is a substrate for p-glycoprotein (P-gp) efflux. Therefore, inhibitors or inducers of these clearance pathways may affect posaconazole plasma concentrations. A summary of drugs studied clinically with the oral suspension or another tablet formulation, which affect posaconazole concentrations, is provided in Table 22.
Table 23 and Table 24 include a summary of the drug effects of concomitant medications that may impact the absorption of posaconazole when administered as either the oral suspension or delayed-release tablets.
A clinical study in healthy volunteers also indicates that posaconazole is a strong CYP3A4 inhibitor as evidenced by a >5-fold increase in midazolam AUC. Therefore, plasma concentrations of drugs predominantly metabolized by CYP3A4 may be increased by posaconazole. A summary of the drugs studied clinically, for which plasma concentrations were affected by posaconazole, is provided in Table 25 [see Contraindications (4) and Drug Interactions (7.2) including recommendations].
Effects of Other Drugs on Posaconazole:
| Coadministered Drug (Postulated Mechanism of Interaction) | Coadministered Drug Dose/Schedule | Noxafil Dose/Schedule | Effect on Bioavailability of Posaconazole | |
|---|---|---|---|---|
| Change in Mean C max (ratio estimate Ratio Estimate is the ratio of coadministered drug plus Noxafil to Noxafil alone for C max or AUC. ; 90% CI of the ratio estimate) | Change in Mean AUC (ratio estimate; 90% CI of the ratio estimate) | |||
| Efavirenz (UDP-G Induction) | 400 mg once daily × 10 and 20 days | 400 mg (oral suspension) twice daily × 10 and 20 days | ↓45% (0.55; 0.47-0.66) | ↓50% (0.50; 0.43-0.60) |
| Fosamprenavir (unknown mechanism) | 700 mg twice daily x 10 days | 200 mg once daily on the 1 st day, 200 mg twice daily on the 2 nd day, then 400 mg twice daily x 8 Days | ↓21% 0.79 (0.71-0.89) | ↓23% 0.77 (0.68-0.87) |
| Rifabutin (UDP-G Induction) | 300 mg once daily x 17 days | 200 mg (tablets) once daily × 10 days The tablet refers to a non-commercial tablet formulation without polymer. | ↓43% (0.57; 0.43-0.75) | ↓49% (0.51; 0.37-0.71) |
| Phenytoin (UDP-G Induction) | 200 mg once daily x 10 days | 200 mg (tablets) once daily × 10 days | ↓41% (0.59; 0.44-0.79) | ↓50% (0.50; 0.36-0.71) |
Posaconazole Oral Suspension: Concomitant administration of Noxafil oral suspension with drugs affecting gastric pH or gastric motility results in lower posaconazole exposure. (see Table 23 .)
| Coadministered Drug (Postulated Mechanism of Interaction) | Coadministered Drug Dose/Schedule | Noxafil Dose/Schedule | Effect on Bioavailability of Posaconazole | |
|---|---|---|---|---|
| Change in Mean C max (ratio estimate Ratio Estimate is the ratio of coadministered drug plus Noxafil to coadministered drug alone for C max or AUC. ; 90% CI of the ratio estimate) | Change in Mean AUC (ratio estimate; 90% CI of the ratio estimate) | |||
| Cimetidine (Alteration of gastric pH) | 400 mg twice daily × 10 days | 200 mg (tablets) once daily × 10 days The tablet refers to a non-commercial tablet formulation without polymer. | ↓39% (0.61; 0.53-0.70) | ↓39% (0.61; 0.54-0.69) |
| Esomeprazole (Increase in gastric pH) The drug interactions associated with the oral suspension are also relevant for the delayed-release tablet with the exception of Esomeprazole and Metoclopramide. | 40 mg every morning × 3 days | 400 mg (oral suspension) single dose | ↓46% (0.54; 0.43-0.69) | ↓32% (0.68; 0.57-0.81) |
| Metoclopramide (Increase in gastric motility) | 10 mg three times a day × 2 days | 400 mg (oral suspension) single dose | ↓21% (0.79; 0.72-0.87) | ↓19% (0.81; 0.72-0.91) |
Posaconazole Delayed-Release Tablets: Concomitant administration of Noxafil delayed-release tablets with drugs affecting gastric pH or gastric motility did not demonstrate any significant effects on posaconazole pharmacokinetic exposure (see Table 24 ).
| Coadministered Drug | Administration Arms | Change in C max (ratio estimate Ratio Estimate is the ratio of coadministered drug plus Noxafil to Noxafil alone for C max or AUC 0-last . ; 90% CI of the ratio estimate) | Change in AUC 0-last (ratio estimate; 90% CI of the ratio estimate) |
|---|---|---|---|
| Mylanta ® Ultimate strength liquid (Increase in gastric pH) | 25.4 mEq/5 mL, 20 mL | ↑6% (1.06; 0.90-1.26)↑ | ↑4% (1.04; 0.90-1.20) |
| Ranitidine (Zantac ® ) (Alteration in gastric pH) | 150 mg (morning dose of 150 mg Ranitidine twice daily) | ↑4% (1.04; 0.88-1.23)↑ | ↓3% (0.97; 0.84-1.12) |
| Esomeprazole (Nexium ® ) (Increase in gastric pH) | 40 mg (every morning for 5 days, Day -4 to 1) | ↑2% (1.02; 0.88-1.17)↑ | ↑5% (1.05; 0.89-1.24) |
| Metoclopramide (Reglan ® ) (Increase in gastric motility) | 15 mg four times daily for 2 days (Day -1 and 1) | ↓14% (0.86, 0.73,1.02) | ↓7% (0.93, 0.803,1.07) |
Effects of Posaconazole on Other Drugs:
| Coadministered Drug (Postulated Mechanism of Interaction is Inhibition of CYP3A4 by posaconazole) | Coadministered Drug Dose/Schedule | Noxafil Dose/ Schedule | Effect on Bioavailability of Coadministered Drugs | |
|---|---|---|---|---|
| Change in Mean C max (ratio estimate Ratio Estimate is the ratio of coadministered drug plus Noxafil to coadministered drug alone for C max or AUC. ; 90% CI of the ratio estimate) | Change in Mean AUC (ratio estimate; 90% CI of the ratio estimate) | |||
| Sirolimus | 2-mg single oral dose | 400 mg (oral suspension) twice daily x 16 days | ↑572% (6.72; 5.62-8.03) | ↑788% (8.88; 7.26-10.9) |
| Cyclosporine | Stable maintenance dose in heart transplant recipients | 200 mg (tablets) once daily x 10 days The tablet refers to a non-commercial tablet formulation without polymer. | ↑ Cyclosporine whole blood trough concentrations Cyclosporine dose reductions of up to 29% were required | |
| Tacrolimus | 0.05-mg/kg single oral dose | 400 mg (oral suspension) twice daily × 7 days | ↑121% (2.21; 2.01-2.42) | ↑358% (4.58; 4.03-5.19) |
| Simvastatin | 40-mg single oral dose | 100 mg (oral suspension) once daily x 13 days | Simvastatin ↑841% (9.41, 7.13-12.44) Simvastatin Acid ↑817% (9.17, 7.36-11.43) | Simvastatin ↑931% (10.31, 8.40-12.67) Simvastatin Acid ↑634% (7.34, 5.82-9.25) |
| 200 mg (oral suspension) once daily x 13 days | Simvastatin ↑1041% (11.41, 7.99-16.29) Simvastatin Acid ↑851% (9.51, 8.15-11.10) | Simvastatin ↑960% (10.60, 8.63-13.02) Simvastatin Acid ↑748% (8.48, 7.04-10.23) | ||
| Midazolam | 0.4-mg single intravenous dose The mean terminal half-life of midazolam was increased from 3 hours to 7 to 11 hours during coadministration with Noxafil. | 200 mg (oral suspension) twice daily x 7 days | ↑30% (1.3; 1.13-1.48) | ↑362% (4.62; 4.02-5.3) |
| 0.4-mg single intravenous dose | 400 mg (oral suspension) twice daily x 7 days | ↑62% (1.62; 1.41-1.86) | ↑524% (6.24; 5.43-7.16) | |
| 2-mg single oral dose | 200 mg (oral suspension) once daily x 7 days | ↑169% (2.69; 2.46-2.93) | ↑470% (5.70; 4.82-6.74) | |
| 2-mg single oral dose | 400 mg (oral suspension) twice daily x 7 days | ↑138% (2.38; 2.13-2.66) | ↑397% (4.97; 4.46-5.54) | |
| Rifabutin | 300 mg once daily x 17 days | 200 mg (tablets) once daily × 10 days | ↑31% (1.31; 1.10-1.57) | ↑72% (1.72;1.51-1.95) |
| Phenytoin | 200 mg once daily PO x 10 days | 200 mg (tablets) once daily x 10 days | ↑16% (1.16; 0.85-1.57) | ↑16% (1.16; 0.84-1.59) |
| Ritonavir | 100 mg once daily x 14 days | 400 mg (oral suspension) twice daily x 7 days | ↑49% (1.49; 1.04-2.15) | ↑80% (1.8;1.39-2.31) |
| Atazanavir | 300 mg once daily x 14 days | 400 mg (oral suspension) twice daily x 7 days | ↑155% (2.55; 1.89-3.45) | ↑268% (3.68; 2.89-4.70) |
| Atazanavir/ritonavir boosted regimen | 300 mg/100 mg once daily x 14 days | 400 mg (oral suspension) twice daily x 7 days | ↑53% (1.53; 1.13-2.07) | ↑146% (2.46; 1.93-3.13) |
Microbiology
Mechanism of Action
Posaconazole blocks the synthesis of ergosterol, a key component of the fungal cell membrane, through the inhibition of cytochrome P-450 dependent enzyme lanosterol 14α-demethylase responsible for the conversion of lanosterol to ergosterol in the fungal cell membrane. This results in an accumulation of methylated sterol precursors and a depletion of ergosterol within the cell membrane thus weakening the structure and function of the fungal cell membrane. This may be responsible for the antifungal activity of posaconazole.
Resistance
Clinical isolates of Candida albicans and Candida glabrata with decreased susceptibility to posaconazole were observed in oral swish samples taken during prophylaxis with posaconazole and fluconazole, suggesting a potential for development of resistance. These isolates also showed reduced susceptibility to other azoles, suggesting cross-resistance between azoles. The clinical significance of this finding is not known.
Antimicrobial Activity:
Posaconazole has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1) ] .
Microorganisms
Aspergillus spp. and Candida spp.
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC .
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No drug-related neoplasms were recorded in rats or mice treated with posaconazole for 2 years at doses higher than the clinical dose. In a 2 years carcinogenicity study, rats were given posaconazole orally at doses up to 20 mg/kg (females), or 30 mg/kg (males). These doses are equivalent to 3.9- or 3.5-times the exposure achieved with a 400 mg twice daily oral suspension regimen, respectively, based on steady-state AUC in healthy volunteers administered a high-fat meal (400 mg twice daily oral suspension regimen). In the mouse study, mice were treated at oral doses up to 60 mg/kg/day or 4.8-times the exposure achieved with a 400 mg twice daily oral suspension regimen.
Mutagenesis
Posaconazole was not genotoxic or clastogenic when evaluated in bacterial mutagenicity (Ames), a chromosome aberration study in human peripheral blood lymphocytes, a Chinese hamster ovary cell mutagenicity study, and a mouse bone marrow micronucleus study.
Impairment of Fertility
Posaconazole had no effect on fertility of male rats at a dose up to 180 mg/kg (1.7 x the 400 mg twice daily oral suspension regimen based on steady-state plasma concentrations in healthy volunteers) or female rats at a dose up to 45 mg/kg (2.2 x the 400 mg twice daily oral suspension regimen).
CLINICAL STUDIES
Treatment of Invasive Aspergillosis with Noxafil Injection and Noxafil Delayed-Release Tablets
Aspergillosis Treatment Study (NCT01782131) was a randomized, double-blind, controlled trial which evaluated the safety and efficacy of Noxafil injection and Noxafil delayed-release tablets versus voriconazole for primary treatment of invasive fungal disease caused by Aspergillus species. Eligible patients had proven, probable, or possible invasive fungal infections per the European Organization for Research and Treatment of Cancer/Mycoses Study Group, EORTC/MSG criteria. Patients were stratified by risk for mortality or poor outcome where high risk included a history of allogeneic bone marrow transplant, liver transplant, or relapsed leukemia undergoing salvage chemotherapy. The median age of patients was 57 years (range: 14-91 years), with 27.8% of patients aged ≥65 years; 5 patients were pediatric patients 14-16 years of age, of whom 3 were treated with Noxafil and 2 with voriconazole. The majority of patients were male (59.8%) and white (67.1%). With regard to risk factors for invasive aspergillosis, approximately two-thirds of the patients in the study had a recent history of neutropenia, while approximately 20% with a history of an allogeneic stem cell transplant. Over 80% of subjects in each treatment group had infection limited to the lower respiratory tract (primarily lung), while approximately 11% to 13% also had infection in another organ. Invasive aspergillosis was proven or probable in 58.1% of patients as classified by independent adjudicators blinded to study treatment assignment. At least one Aspergillus species was identified in 21% of the patients; A. fumigatus and A. flavus were the most common pathogens identified.
Patients randomized to receive Noxafil were given a dose of 300 mg once daily (twice daily on Day 1) IV or tablet. Patients randomized to receive voriconazole were given a dose of 6 mg/kg twice daily Day 1 followed by 4 mg/kg twice daily IV, or oral 300 mg twice daily Day 1 followed by 200 mg twice daily. The recommended initial route of administration was IV; however, patients could begin oral therapy if clinically stable and able to tolerate oral dosing. The transition from IV to oral therapy occurred when the patient was clinically stable. The protocol recommended duration of therapy was 84 days with a maximum allowed duration of 98 days. Median treatment duration was 67 days for Noxafil patients and 64 days for voriconazole patients. Overall, 55% to 60% of patients began treatment with the IV formulation with a median duration of 9 days for the initial IV dosing.
The Intent to Treat (ITT) population included all patients randomized and receiving at least one dose of study treatment. All-cause mortality through Day 42 in the overall population (ITT) was 15.3% for Noxafil patients compared to 20.6% for voriconazole patients for an adjusted treatment difference of -5.3% with a 95% confidence interval of -11.6 to 1.0%. Consistent results were seen in patients with proven or probable invasive aspergillosis per EORTC criteria (see Table 26 ).
| Noxafil Injection and Delayed-Release Tablets | Voriconazole | ||||
|---|---|---|---|---|---|
| Population | N | n (%) | N | n (%) | Difference Adjusted treatment difference based on Miettinen and Nurminen’s method stratified by randomization factor (risk for mortality/poor outcome), using Cochran-Mantel-Haenszel weighting scheme. (95% CI) |
| Intent to Treat | 288 | 44 (15.3) | 287 | 59 (20.6) | -5.3 (-11.6, 1.0) |
| Proven/Probable Invasive Aspergillosis | 163 | 31 (19.0) | 171 | 32 (18.7) | 0.3 (-8.2, 8.8) |
Global clinical response at Week 6 was assessed by a blinded, independent adjudication committee based upon prespecified clinical, radiologic, and mycologic criteria. In the subgroup of patients with proven or probable invasive aspergillosis per EORTC criteria, the global clinical response of success (complete or partial response) at Week 6 was seen in 44.8% for Noxafil-treated patients compared to 45.6% for voriconazole-treated patients (see Table 27 ).
| Posaconazole | Voriconazole | ||||
|---|---|---|---|---|---|
| Population | N | Success | N | Success | Difference Adjusted treatment difference based on Miettinen and Nurminen’s method stratified by randomization factor (risk for mortality/poor outcome), using Cochran-Mantel-Haenszel weighting scheme. (95% CI) |
| Proven/Probable Invasive Aspergillosis | 163 | 73 (44.8) | 171 | 78 (45.6) | -0.6 (-11.2, 10.1) |
Prophylaxis of Aspergillus and Candida Infections with Noxafil Oral Suspension
Two randomized, controlled studies were conducted using Noxafil as prophylaxis for the prevention of invasive fungal infections (IFIs) among patients at high risk due to severely compromised immune systems.
The first study (Noxafil Oral Suspension Study 1) was a randomized, double-blind trial that compared Noxafil oral suspension (200 mg three times a day) with fluconazole capsules (400 mg once daily) as prophylaxis against invasive fungal infections in allogeneic hematopoietic stem cell transplant (HSCT) recipients with Graft versus Host Disease (GVHD). Efficacy of prophylaxis was evaluated using a composite endpoint of proven/probable IFIs, death, or treatment with systemic antifungal therapy (patients may have met more than one of these criteria). This assessed all patients while on study therapy plus 7 days and at 16 weeks post-randomization. The mean duration of therapy was comparable between the 2 treatment groups (80 days, Noxafil oral suspension; 77 days, fluconazole). Table 28 contains the results from Noxafil Oral Suspension Study 1.
| Posaconazole n=301 | Fluconazole n=299 | |
|---|---|---|
| On therapy plus 7 days | ||
| Clinical Failure Patients may have met more than one criterion defining failure. | 50 (17%) | 55 (18%) |
| Failure due to: | ||
| Proven/Probable IFI | 7 (2%) | 22 (7%) |
| ( Aspergillus ) | 3 (1%) | 17 (6%) |
| ( Candida ) | 1 (<1%) | 3 (1%) |
| (Other) | 3 (1%) | 2 (1%) |
| All Deaths | 22 (7%) | 24 (8%) |
| Proven/probable fungal infection prior to death | 2 (<1%) | 6 (2%) |
| SAF Use of systemic antifungal therapy (SAF) criterion is based on protocol definitions (empiric/IFI usage >4 consecutive days). | 27 (9%) | 25 (8%) |
| Through 16 weeks | ||
| Clinical Failure , 95% confidence interval (posaconazole-fluconazole) = (-11.5%, + 3.7%). | 99 (33%) | 110 (37%) |
| Failure due to: | ||
| Proven/Probable IFI | 16 (5%) | 27 (9%) |
| ( Aspergillus ) | 7 (2%) | 21 (7%) |
| ( Candida ) | 4 (1%) | 4 (1%) |
| (Other) | 5 (2%) | 2 (1%) |
| All Deaths | 58 (19%) | 59 (20%) |
| Proven/probable fungal infection prior to death | 10 (3%) | 16 (5%) |
| SAF | 26 (9%) | 30 (10%) |
| Event free lost to follow-up Patients who are lost to follow-up (not observed for 112 days), and who did not meet another clinical failure endpoint. These patients were considered failures. | 24 (8%) | 30 (10%) |
The second study (Noxafil Oral Suspension Study 2) was a randomized, open-label study that compared Noxafil oral suspension (200 mg 3 times a day) with fluconazole suspension (400 mg once daily) or itraconazole oral solution (200 mg twice a day) as prophylaxis against IFIs in neutropenic patients who were receiving cytotoxic chemotherapy for AML or MDS. As in Noxafil Oral Suspension Study 1, efficacy of prophylaxis was evaluated using a composite endpoint of proven/probable IFIs, death, or treatment with systemic antifungal therapy (Patients might have met more than one of these criteria). This study assessed patients while on treatment plus 7 days and 100 days postrandomization. The mean duration of therapy was comparable between the 2 treatment groups (29 days, posaconazole; 25 days, fluconazole or itraconazole). Table 29 contains the results from Noxafil Oral Suspension Study 2.
| Posaconazole n=304 | Fluconazole/Itraconazole n=298 | |
|---|---|---|
| On therapy plus 7 days | ||
| Clinical Failure 95% confidence interval (posaconazole-fluconazole/itraconazole) = (-22.9%, -7.8%). , Patients may have met more than one criterion defining failure. | 82 (27%) | 126 (42%) |
| Failure due to: | ||
| Proven/Probable IFI | 7 (2%) | 25 (8%) |
| ( Aspergillus ) | 2 (1%) | 20 (7%) |
| ( Candida ) | 3 (1%) | 2 (1%) |
| (Other) | 2 (1%) | 3 (1%) |
| All Deaths | 17 (6%) | 25 (8%) |
| Proven/probable fungal infection prior to death | 1 (<1%) | 2 (1%) |
| SAF Use of systemic antifungal therapy (SAF) criterion is based on protocol definitions (empiric/IFI usage >3 consecutive days). | 67 (22%) | 98 (33%) |
| Through 100 days postrandomization | ||
| Clinical Failure | 158 (52%) | 191 (64%) |
| Failure due to: | ||
| Proven/Probable IFI | 14 (5%) | 33 (11%) |
| ( Aspergillus ) | 2 (1%) | 26 (9%) |
| ( Candida ) | 10 (3%) | 4 (1%) |
| (Other) | 2 (1%) | 3 (1%) |
| All Deaths | 44 (14%) | 64 (21%) |
| Proven/probable fungal infection prior to death | 2 (1%) | 16 (5%) |
| SAF | 98 (32%) | 125 (42%) |
| Event free lost to follow-up Patients who are lost to follow-up (not observed for 100 days), and who did not meet another clinical failure endpoint. These patients were considered failures. | 34 (11%) | 24 (8%) |
In summary, 2 clinical studies of prophylaxis were conducted with the Noxafil oral suspension. As seen in the accompanying tables (Tables 23 and 24), clinical failure represented a composite endpoint of breakthrough IFI, mortality and use of systemic antifungal therapy. In Noxafil Oral Suspension Study 1 (Table 23), the clinical failure rate of posaconazole (33%) was similar to fluconazole (37%), (95% CI for the difference posaconazole–comparator -11.5% to 3.7%) while in Noxafil Oral Suspension Study 2 (Table 24) clinical failure was lower for patients treated with posaconazole (27%) when compared to patients treated with fluconazole or itraconazole (42%), (95% CI for the difference posaconazole–comparator -22.9% to -7.8%).
All-cause mortality was similar at 16 weeks for both treatment arms in Noxafil Oral Suspension Study 1 [POS 58/301 (19%) vs. FLU 59/299 (20%)]; all-cause mortality was lower at 100 days for Noxafil-treated patients in Noxafil Oral Suspension Study 2 [POS 44/304 (14%) vs. FLU/ITZ 64/298 (21%)]. Both studies demonstrated fewer breakthrough infections caused by Aspergillus species in patients receiving Noxafil prophylaxis when compared to patients receiving fluconazole or itraconazole.
Treatment of Oropharyngeal Candidiasis with Noxafil Oral Suspension
Noxafil Oral Suspension Study 3 was a randomized, controlled, evaluator-blinded study in HIV-infected patients with oropharyngeal candidiasis. Patients were treated with Noxafil or fluconazole oral suspension (both Noxafil and fluconazole were given as follows: 100 mg twice a day for 1 day followed by 100 mg once a day for 13 days).
Clinical and mycological outcomes were assessed after 14 days of treatment and at 4 weeks after the end of treatment. Patients who received at least 1 dose of study medication and had a positive oral swish culture of Candida species at baseline were included in the analyses (see Table 25 ). The majority of the subjects had C. albicans as the baseline pathogen.
Clinical success at Day 14 (complete or partial resolution of all ulcers and/or plaques and symptoms) and clinical relapse rates (recurrence of signs or symptoms after initial cure or improvement) 4 weeks after the end of treatment were similar between the treatment arms (see Table 30 ).
Mycologic eradication rates (absence of colony forming units in quantitative culture at the end of therapy, Day 14), as well as mycologic relapse rates (4 weeks after the end of treatment) were also similar between the treatment arms (see Table 30 ).
| Noxafil | Fluconazole | |
|---|---|---|
| Clinical Success at End of Therapy (Day 14) | 155/169 (91.7%) | 148/160 (92.5%) |
| Clinical Relapse (4 Weeks after End of Therapy) | 45/155 (29.0%) | 52/148 (35.1%) |
| Mycological Eradication (absence of CFU) at End of Therapy (Day 14) | 88/169 (52.1%) | 80/160 (50.0%) |
| Mycological Relapse (4 Weeks after End of Treatment) | 49/88 (55.6%) | 51/80 (63.7%) |
Mycologic response rates, using a criterion for success as a posttreatment quantitative culture with ≤20 colony forming units (CFU/mL) were also similar between the two groups (Noxafil 68.0%, fluconazole 68.1%). The clinical significance of this finding is unknown.
Noxafil Oral Suspension Treatment of Oropharyngeal Candidiasis Refractory to Treatment with Fluconazole or Itraconazole
Noxafil Oral Suspension Study 4 was a noncomparative study of Noxafil oral suspension in HIV-infected subjects with OPC that was refractory to treatment with fluconazole or itraconazole. An episode of OPC was considered refractory if there was failure to improve or worsening of OPC after a standard course of therapy with fluconazole greater than or equal to 100 mg/day for at least 10 consecutive days or itraconazole 200 mg/day for at least 10 consecutive days and treatment with either fluconazole or itraconazole had not been discontinued for more than 14 days prior to treatment with Noxafil. Of the 199 subjects enrolled in this study, 89 subjects met these strict criteria for refractory infection.
Forty-five subjects with refractory OPC were treated with Noxafil oral suspension 400 mg twice daily for 3 days, followed by 400 mg once daily for 25 days with an option for further treatment during a 3-month maintenance period. Following a dosing amendment, a further 44 subjects were treated with Noxafil 400 mg twice daily for 28 days. The efficacy of Noxafil was assessed by the clinical success (cure or improvement) rate after 4 weeks of treatment. The clinical success rate was 74.2% (66/89). The clinical success rates for both the original and the amended dosing regimens were similar (73.3% and 75.0%, respectively).
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Posaconazole Delayed-Release Tablets
Posaconazole delayed-release tablets are yellow, coated, oblong, debossed with "100" on one side containing 100 mg of posaconazole. Bottles with child-resistant closures of 60 delayed-release tablets (NDC 0254-2045-02).
Posaconazole Oral Suspension
Posaconazole oral suspension is a white, cherry-flavored suspension in 4-ounce (123 mL) amber glass bottles with child-resistant closures containing 105 mL of suspension (40 mg of posaconazole per mL). Supplied with each oral suspension bottle is a plastic dosing spoon calibrated for measuring 2.5-mL and 5-mL doses (NDC 0254-1016-36).
Storage and Handling
Posaconazole Delayed-Release Tablets
Store at 20°C to 25°C (68°F to 77°F), excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Posaconazole Oral Suspension
Store at 25°C (77°F); excursions permitted to 15 to 30°C (59 to 86°F) [see USP Controlled Room Temperature]. DO NOT FREEZE.
Mechanism of Action
Posaconazole is an azole antifungal agent [see Clinical Pharmacology (12.4) ] .