Get your patient on Spravato (Esketamine Hydrochloride)
Spravato patient education
Patient toolkit
Dosage & administration

Spravato prescribing information
WARNING: SEDATION; DISSOCIATION; RESPIRATORY DEPRESSION; ABUSE AND MISUSE; and SUICIDAL THOUGHTS AND BEHAVIORS
WARNING: SEDATION; DISSOCIATION; RESPIRATORY DEPRESSION; ABUSE AND MISUSE; and SUICIDAL THOUGHTS AND BEHAVIORS
See full prescribing information for complete boxed warning.
- Risk for sedation, dissociation, and respiratory depression after administration. Monitor patients for at least two hours after administration. (5.1 , 5.2 , 5.3 )
- Potential for abuse and misuse. Consider the risks and benefits of prescribing SPRAVATO prior to using in patients at higher risk of abuse. Monitor patients for signs and symptoms of abuse and misuse. (5.4 )
- SPRAVATO is only available through a restricted program called the SPRAVATO REMS. (5.5 )
- Increased risk of suicidal thoughts and behaviors in pediatric and young adult patients taking antidepressants. Closely monitor all antidepressant-treated patients for clinical worsening and emergence of suicidal thoughts and behaviors. SPRAVATO is not approved for use in pediatric patients. (5.6 )
Sedation
- Patients are at risk for sedation after administration of SPRAVATO [see Warnings and Precautions (5.1) ].
Dissociation
- Patients are at risk for dissociative or perceptual changes after administration of SPRAVATO [see Warnings and Precautions (5.2) ].
Respiratory Depression
- Respiratory depression has been observed in postmarketing experience [see Warnings and Precautions (5.3) ] .
Because of the risks of sedation, dissociation, and respiratory depression, patients must be monitored for at least 2 hours at each treatment session, followed by an assessment to determine when the patient is considered clinically stable and ready to leave the healthcare setting [see Warnings and Precautions (5.1 , 5.2 , 5.3) ] .
Abuse and Misuse
- SPRAVATO has the potential to be abused and misused. Consider the risks and benefits of prescribing SPRAVATO prior to use in patients at higher risk of abuse. Monitor patients for signs and symptoms of abuse and misuse [see Warnings and Precautions (5.4) ] .
Because of the risks of serious adverse outcomes resulting from sedation, dissociation, respiratory depression, abuse and misuse, SPRAVATO is only available through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the SPRAVATO REMS [see Warnings and Precautions (5.5) ] .
Suicidal Thoughts and Behaviors
Antidepressants increased the risk of suicidal thoughts and behavior in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors. SPRAVATO is not approved for use in pediatric patients [see Warnings and Precautions (5.6) ] .
INDICATIONS AND USAGE
SPRAVATO is indicated for the treatment of:
- Treatment-resistant depression (TRD) in adults as monotherapy or in conjunction with an oral antidepressant
- Depressive symptoms in adults with major depressive disorder (MDD) with acute suicidal ideation or behavior in conjunction with an oral antidepressant
Limitations of Use
- The effectiveness of SPRAVATO in preventing suicide or in reducing suicidal ideation or behavior has not been demonstrated [see Clinical Studies (14.2) ] . Use of SPRAVATO does not preclude the need for hospitalization if clinically warranted, even if patients experience improvement after an initial dose of SPRAVATO.
- SPRAVATO is not approved as an anesthetic agent. The safety and effectiveness of SPRAVATO as an anesthetic agent have not been established.
DOSAGE AND ADMINISTRATION
- Administer SPRAVATO intranasally under the supervision of a healthcare provider. (2.1 )
- Assess blood pressure prior to and after administration. (2.1 )
- TRD: Evidence of therapeutic benefit should be evaluated at the end of the 4-week induction phase to determine need for continued treatment. (2.2 )
- Depressive symptoms in MDD with acute suicidal ideation or behavior : Evidence of therapeutic benefit should be evaluated after 4 weeks to determine need for continued treatment. Treatment beyond 4 weeks has not been systematically evaluated. (2.3 )
- See Full Prescribing Information for recommended dosage. (2.2 , 2.3 )
- See Full Prescribing Information for important administration instructions. (2.4 )
Important Considerations Prior to Initiating and During Therapy
SPRAVATO must be administered under the direct supervision of a healthcare provider. A treatment session consists of nasal administration of SPRAVATO and post-administration observation under supervision.
Respiratory Status Assessment During Treatment
- Monitor patients for changes in respiratory status for at least 2 hours (including pulse oximetry) at each treatment session [see Warnings and Precautions (5.3) ] .
Blood Pressure Assessment Before and After Treatment
- Assess blood pressure prior to dosing with SPRAVATO [see Warnings and Precautions (5.7) ] .
- If baseline blood pressure is elevated (e.g., >140 mmHg systolic, >90 mmHg diastolic), consider the risks of short term increases in blood pressure and benefit of SPRAVATO treatment [see Warnings and Precautions (5.7) ]. Do not administer SPRAVATO if an increase in blood pressure or intracranial pressure poses a serious risk [see Contraindications (4) ] .
- After dosing with SPRAVATO, reassess blood pressure at approximately 40 minutes (which corresponds with the C max ) and subsequently as clinically warranted.
- If blood pressure is decreasing and the patient appears clinically stable for at least two hours, the patient may be discharged at the end of the post-dose monitoring period; if not, continue to monitor [see Warnings and Precautions (5.7) ].
Food and Liquid Intake Recommendations Prior to Administration
Because some patients may experience nausea and vomiting after administration of SPRAVATO [see Adverse Reactions (6.1) ], advise patients to avoid food for at least 2 hours before administration and to avoid drinking liquids at least 30 minutes prior to administration.
Nasal Corticosteroid or Nasal Decongestant
Patients who require a nasal corticosteroid or nasal decongestant on a dosing day should administer these medications at least 1 hour before SPRAVATO [see Clinical Pharmacology (12.3) ].
Treatment-Resistant Depression
The recommended dosage of SPRAVATO for the treatment of TRD in adults as monotherapy or in conjunction with an oral antidepressant is shown in Table 1. Dosage adjustments should be made based on efficacy and tolerability. Evidence of therapeutic benefit should be evaluated at the end of the induction phase to determine need for continued treatment.
| Adults | |
| Induction Phase | Weeks 1 to 4: |
| Administer twice per week | 56 mg or 84 mg |
| Maintenance Phase | Weeks 5 to 8 : |
| Administer once weekly | 56 mg or 84 mg |
| Week 9 and after : | |
| Administer every 2 weeks or once weekly Dosing frequency should be individualized to the least frequent dosing to maintain remission/response. | 56 mg or 84 mg |
Depressive Symptoms in Patients with Major Depressive Disorder with Acute Suicidal Ideation or Behavior
Administer SPRAVATO in conjunction with an oral antidepressant (AD).
The recommended dosage of SPRAVATO for the treatment of depressive symptoms in adults with MDD with acute suicidal ideation or behavior is 84 mg twice per week for 4 weeks. Dosage may be reduced to 56 mg twice per week based on tolerability. After 4 weeks of treatment with SPRAVATO, evidence of therapeutic benefit should be evaluated to determine need for continued treatment. The use of SPRAVATO, in conjunction with an oral antidepressant, beyond 4 weeks has not been systematically evaluated in the treatment of depressive symptoms in patients with MDD with acute suicidal ideation or behavior.
Administration Instructions
SPRAVATO is for nasal use only. The nasal spray device delivers a total of 28 mg of esketamine. To prevent loss of medication, do not prime the device before use. Use 2 devices (for a 56 mg dose) or 3 devices (for an 84 mg dose), with a 5-minute rest between use of each device. Follow these administration instructions and read the Instructions for Use before administration:
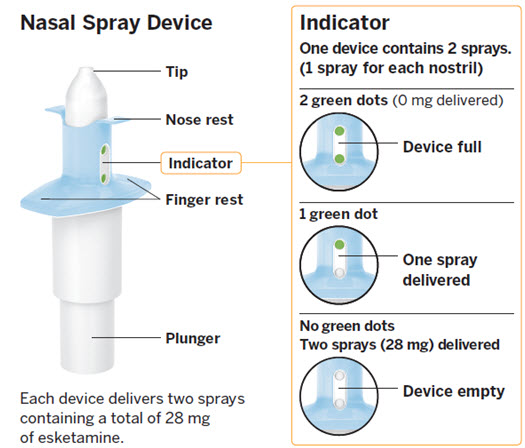

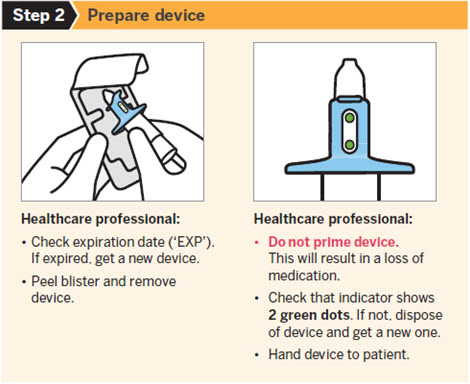
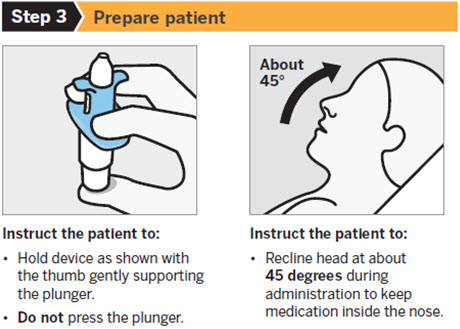

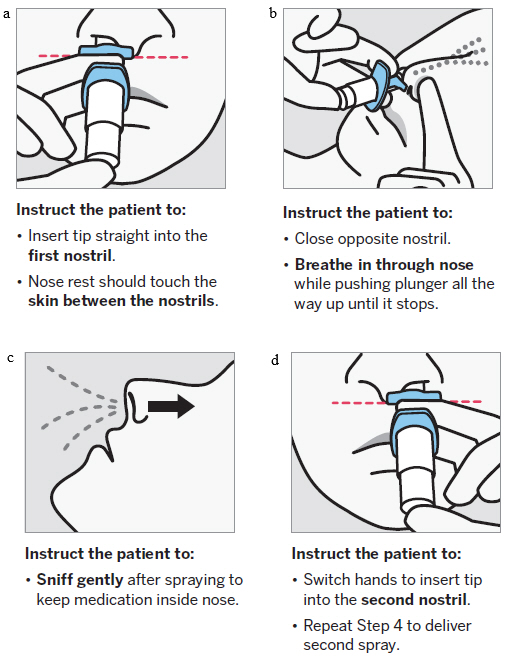
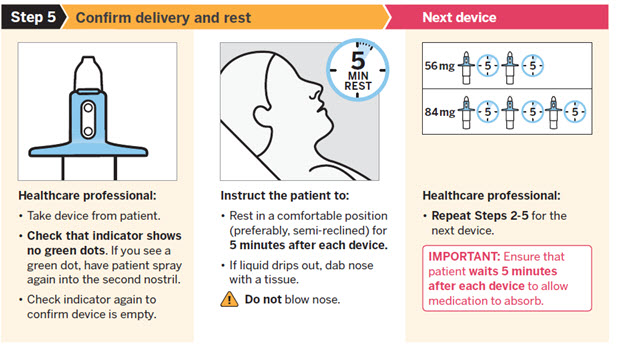

Post-Administration Observation
During and after SPRAVATO administration at each treatment session, observe the patient for at least 2 hours until the patient is safe to leave [see Warnings and Precautions (5.1 , 5.2 , 5.3 , 5.7) ] . Before SPRAVATO administration, instruct patients not to engage in potentially hazardous activities, such as driving a motor vehicle or operating machinery, until the next day after a restful sleep [see Warnings and Precautions (5.9) ] .
Missed Treatment Session(s)
If a patient misses treatment session(s), provided there is no worsening of their depressive symptoms, the patient should continue the current dosing schedule.
For patients who miss treatment session(s) during maintenance treatment and have worsening of depression symptoms, per clinical judgement, consider returning to the previous dosing schedule (e.g., if doses missed during weekly dosing, revert to twice weekly dosing).
DOSAGE FORMS AND STRENGTHS
Nasal Spray: 28 mg of esketamine per device. Each nasal spray device delivers two sprays containing a total of 28 mg esketamine.
USE IN SPECIFIC POPULATIONS
- Lactation: Breastfeeding not recommended. (8.2 )
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antidepressants, including SPRAVATO, during pregnancy. Healthcare providers are encouraged to register patients by contacting the National Pregnancy Registry for Antidepressants at 1-844-405-6185 or online at https://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/antidepressants/ .
Risk Summary
SPRAVATO is not recommended during pregnancy. There are insufficient data on SPRAVATO use in pregnant women to draw conclusions about any drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Based on published findings from pregnant animals treated with ketamine, the racemic mixture of arketamine and esketamine, SPRAVATO may cause fetal harm when administered to pregnant women (see Data ) . Advise pregnant women of the potential risk to an infant exposed to SPRAVATO in utero . There are risks to the mother associated with untreated depression in pregnancy (see Clinical Considerations ). If a woman becomes pregnant while being treated with SPRAVATO, treatment with esketamine should be discontinued and the patient should be counseled about the potential risk to the fetus.
Published studies in pregnant primates demonstrate that the administration of drugs that block N -methyl- D -aspartate (NMDA) receptors during the period of peak brain development increases neuronal apoptosis in the developing brain of the offspring. There are no data on pregnancy exposures in primates corresponding to periods prior to the third trimester in humans [see Use in Specific Populations (8.2) ] .
In an embryo-fetal reproduction study in rabbits, skeletal malformations were noted at maternally toxic doses when ketamine was intranasally administered with a No Observed Adverse Effect Level (NOAEL) at estimated esketamine exposures 0.3 times the exposures at the maximum recommended human dose (MRHD) of 84 mg/day. In addition, intranasal administration of esketamine to pregnant rats during pregnancy and lactation at exposures that were similar to those at the MRHD resulted in a delay in sensorimotor development in pups during the preweaning period and a decrease in motor activity in the post-weaning period.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo-Fetal Risk
A prospective, longitudinal study followed 201 pregnant women with a history of major depressive disorder who were euthymic and taking antidepressants at the beginning of pregnancy. The women who discontinued antidepressants during pregnancy were more likely to experience a relapse of major depression than women who continued antidepressants. Consider the risk of untreated depression when discontinuing or changing treatment with antidepressant medication during pregnancy and postpartum.
Data
Animal Data
Based on published data, when female monkeys were treated intravenously with racemic ketamine at anesthetic dose levels in the third trimester of pregnancy, neuronal cell death was observed in the brains of their fetuses. This period of brain development translates into the third trimester of human pregnancy. The clinical significance of these findings is not clear; however, studies in juvenile animals suggest neuroapoptosis correlates with long-term cognitive deficits.
Racemic ketamine was administered intranasally to pregnant rats during the period of organogenesis at doses of 15, 50, and 150 mg/kg/day. The No Observed Adverse Effect Level (NOAEL) for embryo-fetal toxicity in rats was the highest dose of 150 mg/kg/day. Estimating 50% of the exposure to be from esketamine, the NOAEL associated with esketamine plasma exposure (AUC) is 12-times the AUC exposure at the MRHD of 84 mg/day. In pregnant rabbits, racemic ketamine was administered intranasally from gestational day 6 to 18 at doses of 10, 30, and 100 mg/kg/day. The high dose was lowered from 100 to 50 mg/kg after 5 days of dosing due to excessive mortality in the pregnant rabbits. Skeletal malformations were observed at doses ≥ 30 mg/kg/day, which were maternally toxic. The NOAEL for skeletal malformations was associated with a plasma esketamine exposure (AUC) that was 0.3 times the AUC exposure at MRHD of 84 mg/day.
Administration of esketamine to pregnant rats during pregnancy and lactation at intranasal doses equivalent to 4.5, 15, and 45 mg/kg/day (based on a 200-gram rat) produced AUC exposures 0.07, 0.5, and 0.7 times the MRHD of 84 mg/day, respectively. Maternal toxicity was observed at doses ≥ 15 mg/kg/day. In addition, a dose-dependent delay in the age of attainment of Preyer response reflex was observed in pups at all doses during the preweaning period. This sensory/motor developmental measure was tested starting on postnatal day (PND) 9, and the effect normalized by PND 19 in treatment groups as compared with PND 14 for the majority of the control animals. There is no NOAEL for this delay in sensory/motor response observed in pups during the preweaning period. During the postweaning period, a decrease in motor activity was observed at doses ≥ 15 mg/kg which is 0.5-times the human exposure at the MRHD of 84 mg/day. The NOAEL for maternal toxicity and decreased motor activity during the postweaning period was 4.5 mg/kg/day which was associated with a plasma exposure (AUC) that was 0.07-times the AUC exposure at MRHD of 84 mg/day.
Lactation
Risk Summary
Esketamine is present in human milk. There are no data on the effects of SPRAVATO on the breastfed infant or on milk production. Published studies in juvenile animals report neurotoxicity (see Data) . Because of the potential for neurotoxicity, advise patients that breast-feeding is not recommended during treatment with SPRAVATO.
Data
Published juvenile animal studies demonstrate that the administration of drugs that block NMDA receptors, such as ketamine, during the period of rapid brain growth or synaptogenesis, results in widespread neuronal and oligodendrocyte cell loss in the developing brain and alterations in synaptic morphology and neurogenesis. Based on comparisons across species, the window of vulnerability to these changes is believed to correlate with exposures in the third trimester of gestation through the first several months of life, but this window may extend out to approximately 3 years of age in humans.
Females and Males of Reproductive Potential
Contraception
Based on published animal reproduction studies, SPRAVATO may cause embryo-fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.11) and Use in Specific Populations (8.1) ]. However, it is not clear how these animal findings relate to females of reproductive potential treated with the recommended clinical dose. Consider pregnancy planning and prevention for females of reproductive potential during treatment with SPRAVATO.
Pediatric Use
The safety and effectiveness of SPRAVATO in pediatric patients have not been established.
Geriatric Use
Of the total number of patients in randomized, double-blind, placebo-controlled short-term clinical studies exposed to SPRAVATO, (N=2064), 238 (12%) were 65 years of age and older, and 29 (1%) were 75 years of age and older. No overall differences in the safety profile were observed between patients 65 years of age and older and patients younger than 65 years of age.
The mean esketamine C max and AUC values were higher in elderly patients compared with younger adult patients [see Clinical Pharmacology (12.3) ] .
The efficacy of SPRAVATO for the treatment of TRD in geriatric patients was evaluated in a 4-week, randomized, double-blind study comparing flexibly-dosed intranasal SPRAVATO plus a newly initiated oral antidepressant compared to intranasal placebo plus a newly initiated oral antidepressant in patients ≥ 65 years of age. SPRAVATO was initiated at 28 mg twice weekly and could be titrated to 56 mg or 84 mg administered twice weekly. At the end of four weeks, there was no statistically significant difference between groups on the primary efficacy endpoint of change from baseline to Week 4 on the Montgomery-Åsberg Depression Rating Scale (MADRS).
Hepatic Impairment
The mean esketamine AUC and t 1/2 values were higher in patients with moderate hepatic impairment compared to those with normal hepatic function [see Clinical Pharmacology (12.3) ] . SPRAVATO-treated patients with moderate hepatic impairment may need to be monitored for adverse reactions for a longer period of time.
SPRAVATO has not been studied in patients with severe hepatic impairment (Child-Pugh class C). Use in this population is not recommended [see Clinical Pharmacology (12.3) ].
CONTRAINDICATIONS
SPRAVATO is contraindicated in patients with:
- Aneurysmal vascular disease (including thoracic and abdominal aorta, intracranial, and peripheral arterial vessels) or arteriovenous malformation [see Warnings and Precautions (5.7) ]
- History of intracerebral hemorrhage [see Warnings and Precautions (5.7) ]
- Hypersensitivity to esketamine, ketamine, or any of the excipients.
WARNINGS AND PRECAUTIONS
- Increases in Blood Pressure: Patients with cardiovascular and cerebrovascular conditions and risk factors may be at an increased risk of associated adverse effects. (5.7 )
- Cognitive Impairment: SPRAVATO may impair attention, judgment, thinking, reaction speed and motor skills. (5.8 )
- Impaired Ability to Drive and Operate Machinery: Do not drive or operate machinery until the next day after a restful sleep. (5.9 )
- Embryo-fetal Toxicity: May cause fetal harm. Consider pregnancy planning and prevention in females of reproductive potential. (5.11 , 8.1 , 8.3 )
Sedation
SPRAVATO may cause sedation or loss of consciousness. In some cases, patients may display diminished or less apparent breathing. In clinical trials, 48% to 61% of SPRAVATO-treated patients developed sedation based on the Modified Observer's Assessment of Alertness/Sedation scale (MOAA/S) [see Adverse Reactions (6.1) ] , and 0.3% to 0.4% of SPRAVATO-treated patients experienced loss of consciousness (MOAA/S score of 0).
Because of the possibility of delayed or prolonged sedation, patients must be monitored by a healthcare provider for at least 2 hours at each treatment session, followed by an assessment to determine when the patient is considered clinically stable and ready to leave the healthcare setting [see Dosage and Administration (2.5) ] .
Closely monitor for sedation with concomitant use of SPRAVATO with CNS depressants [see Drug Interactions (7.1) ] .
SPRAVATO is available only through a restricted program under a REMS [see Warnings and Precautions (5.5) ] .
Dissociation
The most common psychological effects of SPRAVATO were dissociative or perceptual changes (including distortion of time, space and illusions), derealization and depersonalization (61% to 84% of SPRAVATO-treated patients developed dissociative or perceptual changes based on the Clinician-Administered Dissociative States Scale) [see Adverse Reactions (6.1) ] . Given its potential to induce dissociative effects, carefully assess patients with psychosis before administering SPRAVATO; treatment should be initiated only if the benefit outweighs the risk.
Because of the risks of dissociation, patients must be monitored by a healthcare provider for at least 2 hours at each treatment session, followed by an assessment to determine when the patient is considered clinically stable and ready to leave the healthcare setting [see Dosage and Administration (2.5) ] .
SPRAVATO is available only through a restricted program under a REMS [see Warnings and Precautions (5.5) ] .
Respiratory Depression
In post marketing experience, respiratory depression was observed with the use of SPRAVATO. In addition, there were rare reports of respiratory arrest [see Adverse Reactions (6.2) ] .
Because of the risks of respiratory depression, patients must be monitored for changes in respiratory status by a healthcare provider for at least 2 hours (including pulse oximetry) at each treatment session, followed by an assessment to determine when the patient is considered clinically stable and ready to leave the healthcare setting [see Dosage and Administration (2.5) ] .
SPRAVATO is available only through a restricted program under a REMS [see Warnings and Precautions (5.5) ] .
Abuse and Misuse
SPRAVATO contains esketamine, a Schedule III controlled substance (CIII), and may be subject to abuse and diversion. Assess each patient's risk for abuse or misuse prior to prescribing SPRAVATO and monitor all patients receiving SPRAVATO for the development of these behaviors or conditions, including drug-seeking behavior, while on therapy. Contact local state professional licensing board or state-controlled substances authority for information on how to prevent and detect abuse or diversion of SPRAVATO. Individuals with a history of drug abuse or dependence are at greater risk; therefore, use careful consideration prior to treatment of individuals with a history of substance use disorder and monitor for signs of abuse or dependence [see Drug Abuse and Dependence (9) ] .
SPRAVATO is available only through a restricted program under a REMS [see Warnings and Precautions (5.5) ] .
SPRAVATO Risk Evaluation and Mitigation Strategy (REMS)
SPRAVATO is available only through a restricted program under a REMS called the SPRAVATO REMS because of the risks of serious adverse outcomes from sedation, dissociation, respiratory depression, abuse and misuse [see Boxed Warning and Warnings and Precautions (5.1 , 5.2 , 5.3 , 5.4) ].
Important requirements of the SPRAVATO REMS include the following:
- Healthcare settings must be certified in the program and ensure that SPRAVATO is:
- Only dispensed and administered in healthcare settings.
- Patients treated in outpatient settings (e.g. medical offices and clinics) must be enrolled in the program.
- Administered by patients under the direct observation of a healthcare provider and that patients are monitored by a healthcare provider for at least 2 hours after administration of SPRAVATO [see Dosage and Administration (2.5) ].
- Pharmacies must be certified in the REMS and must only dispense SPRAVATO to healthcare settings that are certified in the program.
Further information, including a list of certified pharmacies is available at www.SPRAVATOrems.com or 1-855-382-6022.
Suicidal Thoughts and Behaviors in Adolescents and Young Adults
In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and 4,500 pediatric patients (SPRAVATO is not approved in pediatric patients), the incidence of suicidal thoughts and behaviors in patients age 24 years and younger was greater than in placebo-treated patients. There was considerable variation in risk of suicidal thoughts and behaviors among drugs, but there was an increased risk identified in young patients for most drugs studied. There were differences in absolute risk of suicidal thoughts and behaviors across the different indications, with the highest incidence in patients with major depressive disorder (MDD). The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1000 patients treated are provided in Table 2.
| Age Range (Years) | Drug-Placebo Difference in Number of Patients with Suicidal Thoughts or Behaviors per 1000 Patients Treated |
|---|---|
| Increases Compared to Placebo | |
| <18 | 14 additional patients |
| 18–24 | 5 additional patients |
| Decreases Compared to Placebo | |
| 25–64 | 1 fewer patient |
| ≥65 | 6 fewer patients |
It is unknown whether the risk of suicidal thoughts and behaviors in children, adolescents, and young adults extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance studies in adults with MDD that antidepressants delay the recurrence of depression and that depression itself is a risk factor for suicidal thoughts and behaviors.
Monitor all antidepressant-treated patients for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of drug therapy and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behavior and to alert the healthcare provider. Consider changing the therapeutic regimen, including possibly discontinuing SPRAVATO and/or the concomitant oral antidepressant, in patients whose depression is persistently worse, or who are experiencing emergent suicidal thoughts or behaviors.
Increase in Blood Pressure
SPRAVATO causes increases in systolic and/or diastolic blood pressure (BP) at all recommended doses. Increases in BP peak approximately 40 minutes after SPRAVATO administration and last approximately 4 hours [see Adverse Reactions (6.1) ] .
Approximately 3% to 19% of SPRAVATO-treated patients and 1% to 4% of placebo-treated patients experienced an increase of greater than or equal to 40 mmHg in systolic BP and/or 25 mmHg in diastolic BP in the first 1.5 hours after administration at least once during the first 4 weeks of treatment. A substantial increase in blood pressure could occur after any dose administered even if smaller blood pressure effects were observed with previous administrations. SPRAVATO is contraindicated in patients for whom an increase in BP or intracranial pressure poses a serious risk (e.g., aneurysmal vascular disease, arteriovenous malformation, history of intracerebral hemorrhage) [see Contraindications (4) ]. Before prescribing SPRAVATO, patients with other cardiovascular and cerebrovascular conditions should be carefully assessed to determine whether the potential benefits of SPRAVATO outweigh its risks.
Assess BP prior to administration of SPRAVATO. In patients whose BP is elevated prior to SPRAVATO administration (as a general guide: >140/90 mmHg) a decision to delay SPRAVATO therapy should take into account the balance of benefit and risk in individual patients.
BP should be monitored for at least 2 hours after SPRAVATO administration [see Dosage and Administration (2.1 , 2.5) ] . Measure blood pressure around 40 minutes post-dose and subsequently as clinically warranted until values decline. If BP remains high, promptly seek assistance from practitioners experienced in BP management. Refer patients experiencing symptoms of a hypertensive crisis (e.g., chest pain, shortness of breath) or hypertensive encephalopathy (e.g., sudden severe headache, visual disturbances, seizures, diminished consciousness or focal neurological deficits) immediately for emergency care.
Closely monitor blood pressure with concomitant use of SPRAVATO with psychostimulants or monoamine oxidase inhibitors (MAOIs) [see Drug Interactions (7.2 , 7.3) ] .
In patients with history of hypertensive encephalopathy, more intensive monitoring, including more frequent blood pressure and symptom assessment, is warranted because these patients are at increased risk for developing encephalopathy with even small increases in blood pressure.
Cognitive Impairment
Short-Term Cognitive Impairment
In a study in healthy volunteers, a single dose of SPRAVATO caused cognitive performance decline 40 minutes post-dose. Compared to placebo-treated subjects, SPRAVATO-treated subjects required a greater effort to complete cognitive tests at 40 minutes post-dose. Cognitive performance and mental effort were comparable between SPRAVATO and placebo at 2 hours post-dose. Sleepiness was comparable after 4 hours post-dose.
Long-Term Cognitive Impairment
Long-term cognitive and memory impairment have been reported with repeated ketamine misuse or abuse. In 1-year and 3-year, long-term, open-label clinical trials in adults, the effect of SPRAVATO on cognitive functioning remained stable over time as evaluated by the Cogstate computerized battery and Hopkins Verbal Learning Test-Revised.
Impaired Ability to Drive and Operate Machinery
Two placebo-controlled studies were conducted to assess the effects of SPRAVATO on the ability to drive [see Clinical Studies (14.3) ] . The effects of SPRAVATO 84 mg were comparable to placebo at 6 hours and 18 hours post-dose. However, two SPRAVATO-treated subjects in one of the studies discontinued the driving test at 8 hours post-dose because of SPRAVATO-related adverse reactions.
Before SPRAVATO administration, instruct patients not to engage in potentially hazardous activities requiring complete mental alertness and motor coordination, such as driving a motor vehicle or operating machinery, until the next day following a restful sleep. Patients will need to arrange transportation home following treatment with SPRAVATO.
Ulcerative or Interstitial Cystitis
Cases of ulcerative or interstitial cystitis have been reported in individuals with long-term off-label use or misuse/abuse of ketamine. In clinical studies with SPRAVATO nasal spray, there was a higher rate of lower urinary tract symptoms (pollakiuria, dysuria, micturition urgency, nocturia, and cystitis) in SPRAVATO-treated patients than in placebo-treated patients [see Adverse Reactions (6) ] . No cases of esketamine-related interstitial cystitis were observed in any of the studies, which included treatment for up to a year.
Monitor for urinary tract and bladder symptoms during the course of treatment with SPRAVATO, and refer to an appropriate healthcare provider as clinically warranted.
Embryo-fetal Toxicity
Based on published findings from pregnant animals treated with ketamine, the racemic mixture of arketamine and esketamine, SPRAVATO may cause fetal harm when administered to pregnant women. Advise pregnant women of the potential risk to an infant exposed to SPRAVATO in utero . Advise women of reproductive potential to consider pregnancy planning and prevention [see Use in Specific Populations (8.1 , 8.3) ].
ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Sedation [see Warnings and Precautions (5.1) ]
- Dissociation [see Warnings and Precautions (5.2) ]
- Respiratory Depression [see Warnings and Precautions (5.3) ]
- Increase in Blood Pressure [see Warnings and Precautions (5.7) ]
- Cognitive Impairment [see Warnings and Precautions (5.8) ]
- Impaired Ability to Drive and Operate Machinery [see Warnings and Precautions (5.9) ]
- Ulcerative or Interstitial Cystitis [see Warnings and Precautions (5.10) ]
- Embryo-fetal Toxicity [see Warnings and Precautions (5.11) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Treatment-Resistant Depression
In Conjunction with an Oral Antidepressant
SPRAVATO was evaluated for safety in 1709 adults diagnosed with treatment-resistant depression (TRD) [see Clinical Studies (14.1) ] from five Phase 3 studies (3 short-term and 2 long-term studies) and one Phase 2 dose-ranging study. Of all SPRAVATO-treated patients in the completed Phase 3 studies, 479 (30%) received at least 6 months of treatment, and 178 (11%) received at least 12 months of treatment.
Adverse Reactions Leading to Discontinuation of Treatment
In short-term studies in adults <65 years old (Study 1 pooled with another 4-week study), the proportion of patients who discontinued treatment because of an adverse reaction was 4.6% in patients who received SPRAVATO plus oral AD compared to 1.4% for patients who received placebo nasal spray plus oral AD. For adults ≥65 years old, the proportions were 5.6% and 3.1%, respectively. In Study 2, a long-term maintenance study, the discontinuation rates because of an adverse reaction were similar for patients receiving SPRAVATO plus oral AD and placebo nasal spray plus oral AD in the maintenance phase, at 2.6% and 2.1%, respectively. Across all Phase 3 studies, adverse reactions leading to SPRAVATO discontinuation in more than 2 patients were (in order of frequency): anxiety (1.2%), depression (0.9%), blood pressure increased (0.6%), dizziness (0.6%), suicidal ideation (0.5%), dissociation (0.4%), nausea (0.4%), vomiting (0.4%), headache (0.3%), muscular weakness (0.3%), vertigo (0.2%), hypertension (0.2%), panic attack (0.2%) and sedation (0.2%).
Most Common Adverse Reactions
The most commonly observed adverse reactions in patients treated with SPRAVATO plus oral AD (incidence ≥5% and at least twice that of placebo nasal spray plus oral AD) were dissociation, dizziness, nausea, sedation, vertigo, hypoesthesia, anxiety, lethargy, blood pressure increased, vomiting, and feeling drunk.
Table 3 shows the incidence of adverse reactions that occurred in patients treated with SPRAVATO plus oral AD at any dose and greater than patients treated with placebo nasal spray plus oral AD.
| SPRAVATO + Oral AD (N=346) | Placebo + Oral AD (N=222) | |
|---|---|---|
| Cardiac disorders | ||
| Tachycardia The following terms were combined: Anxiety includes: agitation; anticipatory anxiety; anxiety; fear; feeling jittery; irritability; nervousness; panic attack; tension Blood pressure increased includes: blood pressure diastolic increased; blood pressure increased; blood pressure systolic increased; hypertension Dissociation includes: delusional perception; depersonalization/derealization disorder; derealization; diplopia; dissociation; dysesthesia; feeling cold; feeling hot; feeling of body temperature change; hallucination; hallucination, auditory; hallucination, visual; hyperacusis; illusion; ocular discomfort; oral dysesthesia; paresthesia; paresthesia oral; pharyngeal paresthesia; photophobia; time perception altered; tinnitus; vision blurred; visual impairment Dizziness includes: dizziness; dizziness exertional; dizziness postural; procedural dizziness Dysarthria includes: dysarthria; slow speech; speech disorder Dysgeusia includes: dysgeusia; hypogeusia Headache includes: headache; sinus headache Hypoesthesia includes: hypoesthesia; hypoesthesia oral, hypoesthesia teeth, pharyngeal hypoesthesia Lethargy includes: fatigue; lethargy Nasal discomfort includes: nasal crusting; nasal discomfort; nasal dryness; nasal pruritus Sedation includes: altered state of consciousness; hypersomnia; sedation; somnolence Tachycardia includes: extrasystoles; heart rate increased; tachycardia Vertigo includes: vertigo; vertigo positional | 6 (2%) | 1 (0.5%) |
| Ear and labyrinth disorders | ||
| Vertigo | 78 (23%) | 6 (3%) |
| Gastrointestinal disorders | ||
| Nausea | 98 (28%) | 19 (9%) |
| Vomiting | 32 (9%) | 4 (2%) |
| Diarrhea | 23 (7%) | 13 (6%) |
| Dry mouth | 19 (5%) | 7 (3%) |
| Constipation | 11 (3%) | 3 (1%) |
| General disorders and administration site conditions | ||
| Feeling drunk | 19 (5%) | 1 (0.5%) |
| Feeling abnormal | 12 (3%) | 0 (0%) |
| Investigations | ||
| Blood pressure increased | 36 (10%) | 6 (3%) |
| Nervous system disorders | ||
| Dizziness | 101 (29%) | 17 (8%) |
| Sedation | 79 (23%) | 21 (9%) |
| Headache | 70 (20%) | 38 (17%) |
| Dysgeusia | 66 (19%) | 30 (14%) |
| Hypoesthesia | 63 (18%) | 5 (2%) |
| Lethargy | 37 (11%) | 12 (5%) |
| Dysarthria | 15 (4%) | 0 (0%) |
| Tremor | 12 (3%) | 2 (1%) |
| Mental impairment | 11 (3%) | 2 (1%) |
| Psychiatric disorders | ||
| Dissociation | 142 (41%) | 21 (9%) |
| Anxiety | 45 (13%) | 14 (6%) |
| Insomnia | 29 (8%) | 16 (7%) |
| Euphoric mood | 15 (4%) | 2 (1%) |
| Renal and urinary disorders | ||
| Pollakiuria | 11 (3%) | 1 (0.5%) |
| Respiratory, thoracic and mediastinal disorders | ||
| Nasal discomfort | 23 (7%) | 11 (5%) |
| Throat irritation | 23 (7%) | 9 (4%) |
| Oropharyngeal pain | 9 (3%) | 5 (2%) |
| Skin and subcutaneous tissue disorders | ||
| Hyperhidrosis | 14 (4%) | 5 (2%) |
Monotherapy
SPRAVATO used as monotherapy was evaluated for safety in 463 adults diagnosed with treatment-resistant depression (TRD) [see Clinical Studies (14.1) ] .
Adverse Reactions Leading to Discontinuation of Treatment
In the double-blind treatment phase of the short-term study in adults (Study 3), the proportion of patients who discontinued treatment because of an adverse reaction was 1% in patients who received SPRAVATO 56 mg and 4% in patients who received SPRAVATO 84 mg compared to 1% for patients who received placebo nasal spray. All of the adverse reactions leading to SPRAVATO discontinuation occurred in single subjects.
Most Common Adverse Reactions
The most common adverse reactions (≥5% and at least twice that of placebo nasal spray) were dissociation, nausea, dizziness, headache, anxiety, vomiting, feeling drunk, blood pressure increased, and sedation. Table 4 shows the incidence of adverse reactions that occurred in patients treated with SPRAVATO and greater than patients treated with placebo nasal spray.
| SPRAVATO 56mg + 84mg (N=226) | Placebo (N=250) | |
|---|---|---|
| Ear and labyrinth disorders | ||
| Vertigo | 6 (3%) | 0 (0%) |
| Gastrointestinal disorders | ||
| Nausea | 56 (25%) | 21 (8%) |
| Vomiting | 15 (7%) | 1 (<1%) |
| General disorders and administration site conditions | ||
| Feeling drunk | 16 (7%) | 2 (<1%) |
| Investigations | ||
| Blood pressure increased The following terms were combined: Anxiety: anxiety, agitation, fear, feeling jittery, irritability, panic attack Blood pressure increased: blood pressure increased, hypertension Dissociation: depersonalization/derealization disorder, derealization, diplopia, dissociation, feeling hot, paresthesia, paresthesia oral, pharyngeal paresthesia, photophobia, tinnitus, vision blurred Dizziness: dizziness, dizziness postural Hypoesthesia: hypoesthesia, hypoesthesia oral, pharyngeal hypoesthesia Lethargy: fatigue, lethargy Sedation: sedation, somnolence | 11 (5%) | 4 (2%) |
| Nervous system disorders | ||
| Disturbance in attention | 5(2%) | 0 (0%) |
| Dizziness | 50 (22%) | 18 (7%) |
| Sedation | 13 (6%) | 4 (2%) |
| Headache | 43 (19%) | 22 (9%) |
| Dysgeusia | 10 (4%) | 9 (4%) |
| Hypoesthesia | 10 (4%) | 1 (<1%) |
| Lethargy | 16 (7%) | 12 (5%) |
| Psychiatric disorders | ||
| Dissociation | 63 (28%) | 11 (4%) |
| Anxiety | 22 (10%) | 8 (3%) |
| Insomnia | 11 (5%) | 9 (4%) |
| Respiratory, thoracic, and mediastinal disorders | ||
| Throat irritation | 8 (4%) | 2 (1%) |
Depressive Symptoms in Patients with Major Depressive Disorder with Acute Suicidal Ideation or Behavior
SPRAVATO was evaluated for safety in 262 adults for the treatment of depressive symptoms in adults with major depressive disorder (MDD) with acute suicidal ideation or behavior [see Clinical Studies (14.2) ] from two Phase 3 studies (Study 4 and Study 5) and one Phase 2 study. Of all SPRAVATO-treated patients in the completed Phase 3 studies, 184 (81%) received all eight doses over a 4-week treatment period.
Adverse Reactions Leading to Discontinuation of Treatment
In short-term studies in adults (pooled Study 4 and Study 5), the proportion of patients who discontinued treatment because of an adverse reaction was 6.2% for patients who received SPRAVATO plus oral AD compared to 3.6% for patients who received placebo nasal spray plus oral AD. Adverse reactions leading to SPRAVATO discontinuation in more than 1 patient were (in order of frequency): dissociation-related events (2.6%), blood pressure increased (0.9%), dizziness-related events (0.9%), nausea (0.9%), and sedation-related events (0.9%).
Most Common Adverse Reactions
The most commonly observed adverse reactions in patients treated with SPRAVATO plus oral AD (incidence ≥5% and at least twice that of placebo nasal spray plus oral AD) were dissociation, dizziness, sedation, blood pressure increased, hypoesthesia, vomiting, euphoric mood, and vertigo. Table 5 shows the incidence of adverse reactions that occurred in patients treated with SPRAVATO plus oral AD and greater than patients treated with placebo nasal spray plus oral AD.
| SPRAVATO + Oral AD (N=227) | Placebo + Oral AD (N=225) | |
|---|---|---|
| Cardiac disorders | ||
| Tachycardia The following terms were combined: Anxiety includes: agitation; anxiety; anxiety disorder; fear; irritability; nervousness; panic attack; psychomotor hyperactivity; tension Blood pressure increased includes: blood pressure diastolic increased; blood pressure increased; blood pressure systolic increased; hypertension Dissociation includes: depersonalization/derealization disorder; derealization; diplopia; dissociation; dysesthesia; feeling cold; feeling hot; hallucination; hallucination, auditory; hallucination, visual; hallucinations, mixed; hyperacusis; paresthesia; paresthesia oral; pharyngeal paresthesia; photophobia; time perception altered; tinnitus; vision blurred Dizziness includes: dizziness; dizziness exertional; dizziness postural Dysgeusia includes: dysgeusia; hypogeusia Hyperhidrosis includes: cold sweat; hyperhidrosis Hypoesthesia includes: hypoesthesia; hypoesthesia oral; intranasal hypoesthesia; pharyngeal hypoesthesia Lethargy includes: fatigue; lethargy; psychomotor retardation Pollakiuria includes: micturition urgency; pollakiuria Sedation includes: sedation; somnolence; stupor Tachycardia includes: heart rate increased; sinus tachycardia; tachycardia | 8 (4%) | 2 (1%) |
| Ear and labyrinth disorders | ||
| Vertigo | 14 (6%) | 1 (0.4%) |
| Gastrointestinal disorders | ||
| Nausea | 61 (27%) | 31 (14%) |
| Vomiting | 26 (11%) | 12 (5%) |
| Constipation | 22 (10%) | 14 (6%) |
| Dry mouth | 8 (4%) | 6 (3%) |
| Toothache | 5 (2%) | 2 (1%) |
| General disorders and administration site conditions | ||
| Feeling drunk | 8 (4%) | 1 (0.4%) |
| Feeling of relaxation | 5 (2%) | 3 (1%) |
| Investigations | ||
| Blood pressure increased | 34 (15%) | 14 (6%) |
| Musculoskeletal and connective tissue disorders | ||
| Myalgia | 5 (2%) | 1 (0.4%) |
| Nervous system disorders | ||
| Dizziness | 103 (45%) | 34 (15%) |
| Sedation | 66 (29%) | 27 (12%) |
| Dysgeusia | 46 (20%) | 29 (13%) |
| Hypoesthesia | 30 (13%) | 4 (2%) |
| Lethargy | 10 (4%) | 4 (2%) |
| Confusional state | 5 (2%) | 0 (0%) |
| Psychiatric disorders | ||
| Dissociation | 108 (48%) | 30 (13%) |
| Anxiety | 34 (15%) | 20 (9%) |
| Euphoric mood | 17 (7%) | 1 (0.4%) |
| Intentional self-injury | 7 (3%) | 3 (1%) |
| Dysphoria | 5 (2%) | 0 (0%) |
| Renal and urinary disorders | ||
| Pollakiuria | 5 (2%) | 2 (1%) |
| Respiratory, thoracic and mediastinal disorders | ||
| Oropharyngeal pain | 10 (4%) | 3 (1%) |
| Throat irritation | 9 (4%) | 5 (2%) |
| Skin and subcutaneous tissue disorders | ||
| Hyperhidrosis | 11 (5%) | 5 (2%) |
Sedation
Sedation was evaluated by adverse event reports and the Modified Observer's Assessment of Alertness/Sedation (MOAA/S). In the MOAA/S, 5 means "responds readily to name spoken in normal tone" and 0 means "no response after painful trapezius squeeze." Any decrease in MOAA/S from pre-dose is considered to indicate the presence of sedation, and such a decrease occurred in a higher number of patients on SPRAVATO than placebo during the short-term TRD studies. Dose-related increases in the incidence of sedation (MOAA/S score <5) were observed in a fixed-dose TRD study [see Warnings and Precautions (5.1) ]. Table 6 presents the incidence of sedation (MOAA/S score <5) in a fixed-dose study with adult patients <65 years of age with TRD and a flexible-dose study with patients ≥65 years of age with TRD.
| Patients <65 years | Patients ≥65 years | ||||
|---|---|---|---|---|---|
| Placebo + Oral AD | SPRAVATO + Oral AD | Placebo + Oral AD | SPRAVATO + Oral AD | ||
| 56 mg | 84 mg | 28 to 84 mg | |||
| Number of patients Patients who were evaluated with MOAA/S | N=112 | N=114 | N=114 | N=63 | N=72 |
| Sedation (MOAA/S score <5) | 11% | 50% | 61% | 19% | 49% |
In studies for the treatment of depressive symptoms in adults with MDD with acute suicidal ideation or behavior, there was a higher incidence of sedation (MOAA/S score <5) in patients treated with SPRAVATO plus oral AD compared to patients treated with placebo plus oral AD, similar to the TRD study results in Table 5.
Dissociation/Perceptual Changes
SPRAVATO can cause dissociative symptoms (including derealization and depersonalization) and perceptual changes (including distortion of time and space, and illusions). In clinical trials, dissociation was transient and occurred on the day of dosing. Dissociation was evaluated by adverse event reports and the Clinician-Administered Dissociative States Scale (CADSS). A CADSS total score of more than 4 indicates the presence of dissociative symptoms, and such an increase to a score of 4 or more occurred in a higher number of patients on SPRAVATO compared to placebo during the short-term TRD studies. Dose-related increases in the incidence of dissociative symptoms (CADSS total score >4 and change >0) were observed in a fixed-dose TRD study [see Warnings and Precautions (5.2) ] . Table 7 presents the incidence of dissociation (CADSS total score >4 and change >0) in a fixed-dose study with adult patients <65 years of age with TRD and a flexible-dose study with patients ≥65 years of age with TRD.
| Patients <65 years | Patients ≥65 years | ||||
|---|---|---|---|---|---|
| Placebo + Oral AD | SPRAVATO + Oral AD | Placebo + Oral AD | SPRAVATO + Oral AD | ||
| 56 mg | 84 mg | 28 to 84 mg | |||
| Number of patients Number of patients who were evaluated with CADSS | N=113 | N=113 | N=116 | N=65 | N=72 |
| CADSS total score >4 and change >0 | 5% | 61% | 69% | 12% | 75% |
In studies for the treatment of depressive symptoms in adults with MDD with acute suicidal ideation or behavior, patients treated with SPRAVATO plus oral AD also demonstrated a higher number (84%) with dissociation (CADSS total score >4 and change >0) compared to patients treated with placebo plus oral AD (16%).
Increase in Blood Pressure
The mean placebo-adjusted increases in systolic and diastolic blood pressure (SBP and DBP) over time were about 7 to 10 mmHg in SBP and 4 to 6 mmHg in DBP at 40 minutes post-dose and 2 to 5 mmHg in SBP and 1 to 3 mmHg in DBP at 1.5 hours post-dose in patients with TRD receiving SPRAVATO [see Warnings and Precautions (5.7) ] . Table 8 presents increases in blood pressure in short-term trials with patients <65 years of age and ≥65 years of age with TRD.
| SPRAVATO + Oral AD | SPRAVATO Monotherapy | |||||
|---|---|---|---|---|---|---|
| Patients <65 years | Patients ≥65 years | Adult patients including those ≥65 years | ||||
| SPRAVATO + Oral AD N=346 | Placebo + Oral AD N=222 | SPRAVATO + Oral AD N=72 | Placebo + Oral AD N=65 | SPRAVATO N=226 | Placebo N=250 | |
| Systolic blood pressure | ||||||
| ≥180 mmHg | 9 (3%) | --- | 2 (3%) | 1 (2%) | 2 (0.9%) | 0 |
| ≥40 mmHg increase | 29 (8%) | 1 (0.5%) | 12 (17%) | 1 (2%) | 18 (8%) | 5 (2%) |
| Diastolic blood pressure | ||||||
| ≥110 mmHg | 13 (4%) | 1 (0.5%) | --- | --- | 3 (1%) | 0 |
| ≥25 mmHg increase | 46 (13%) | 6 (3%) | 10 (14%) | 2 (3%) | 25 (11%) | 6 (2%) |
In studies for the treatment of depressive symptoms in adults with MDD with acute suicidal ideation or behavior, patients treated with SPRAVATO plus oral antidepressants demonstrated similar mean placebo-adjusted increases in SBP and DBP compared to patients with TRD, as well as similar rates of increases to SBP ≥180 mmHg or ≥40 mmHg increases in SBP, and similar rates of increases to DBP ≥110 mmHg or ≥25 mmHg increases in DBP, compared to the TRD study results in Table 8.
Nausea and Vomiting
SPRAVATO can cause nausea and vomiting. Most of these events occurred on the day of dosing and resolved the same day, with the median duration not exceeding 1 hour in most subjects across dosing sessions. Rates of reported nausea and vomiting decreased over time across dosing sessions from the first week of treatment in the short-term studies, as well as over time with long-term treatment. Table 9 presents the incidence and severity of nausea and vomiting in a short-term study with patients with TRD.
| Nausea | Vomiting | ||||
|---|---|---|---|---|---|
| Treatment (+ Oral AD) | |||||
| N | All | Severe | All | Severe | |
| SPRAVATO 56 mg | 115 | 31 (27%) | 0 | 7 (6%) | 0 |
| SPRAVATO 84 mg | 116 | 37 (32%) | 4 (3%) | 14 (12%) | 3 (3%) |
| Placebo Nasal Spray | 113 | 12 (11%) | 0 | 2 (2%) | 0 |
In studies for the treatment of depressive symptoms in adults with MDD with acute suicidal ideation or behavior, patients demonstrated similar incidence and severity of reported nausea and vomiting compared to the TRD study results described above.
Sense of Smell
Sense of smell was assessed over time; no difference was observed between patients treated with SPRAVATO plus oral AD and those treated with placebo nasal spray plus oral AD during the double-blind maintenance phase of Study 2 [see Clinical Studies (14.1) ] .
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of SPRAVATO. Because these reactions are reported voluntarily, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac disorders: bradycardia
Respiratory, thoracic and mediastinal disorders: respiratory depression (including respiratory arrest)
Vascular disorders: hypotension
DRUG INTERACTIONS
Central Nervous System Depressants
Concomitant use with CNS depressants (e.g., benzodiazepines, opioids, alcohol) may increase sedation [see Warnings and Precautions (5.1) ] . Closely monitor for sedation with concomitant use of SPRAVATO with CNS depressants.
Psychostimulants
Concomitant use with psychostimulants (e.g., amphetamines, methylphenidate, modafinil, armodafinil) may increase blood pressure [see Warnings and Precautions (5.7) ] . Closely monitor blood pressure with concomitant use of SPRAVATO with psychostimulants.
Monoamine Oxidase Inhibitors (MAOIs)
Concomitant use with monoamine oxidase inhibitors (MAOIs) may increase blood pressure [see Warnings and Precautions (5.7) ] . Closely monitor blood pressure with concomitant use of SPRAVATO with MAOIs.
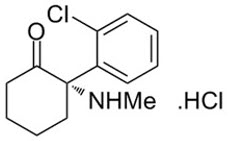
DESCRIPTION
SPRAVATO ® contains esketamine hydrochloride, a non-competitive N -methyl- D -aspartate (NMDA) receptor antagonist. Esketamine is the S-enantiomer of racemic ketamine. The chemical name is (S) -2-(o-chlorophenyl)-2-(methylamino)cyclohexanone hydrochloride. Its molecular formula is C 13 H 16 ClNO.HCl and its molecular weight is 274.2. The structural formula is:
Esketamine hydrochloride is a white or almost white crystalline powder that is freely soluble in water and in methanol, and soluble in ethanol.
SPRAVATO nasal spray is intended for nasal administration. Esketamine hydrochloride is contained as a solution in a stoppered glass vial within the nasal spray device. Each device delivers two sprays with a total of 32.3 mg of esketamine hydrochloride (equivalent to 28 mg of esketamine) in 0.2 mL of a clear, colorless aqueous solution with a pH of 4.5.
The inactive ingredients are citric acid monohydrate, edetate disodium, sodium hydroxide, and water for injection.
CLINICAL PHARMACOLOGY
Mechanism of Action
Esketamine, the S-enantiomer of racemic ketamine, is a non-selective, non-competitive antagonist of the N -methyl- D -aspartate (NMDA) receptor, an ionotropic glutamate receptor. The mechanism by which esketamine exerts its antidepressant effect is unknown. The major circulating metabolite of esketamine (noresketamine) demonstrated activity at the same receptor with less affinity.
Pharmacodynamics
Cardiac Electrophysiology
The effect of SPRAVATO (84 mg nasal spray and 0.8 mg/kg esketamine intravenously infused over 40 minutes) on the QTc interval was evaluated in a randomized, double-blind, placebo-, and positive-controlled (moxifloxacin 400 mg), 4-period, crossover study in 60 healthy subjects. A large increase in heart rate (i.e., >10 bpm) was observed in both intranasal and intravenous esketamine treatment groups. The totality of evidence from the nonclinical and clinical data indicates a lack of clinically relevant QTc prolongation at the therapeutic dose of esketamine.
Pharmacokinetics
Esketamine exposure increases with dose from 28 mg to 84 mg. The increase in C max and AUC values was less than dose-proportional between 28 mg and 56 mg or 84 mg, but it was nearly dose proportional between 56 mg and 84 mg. No accumulation of esketamine in plasma was observed following twice a week administration.
Absorption
The mean absolute bioavailability is approximately 48% following nasal spray administration.
The time to reach maximum esketamine plasma concentration is 20 to 40 minutes after the last nasal spray of a treatment session.
The inter-subject variability of esketamine ranges from 27% to 66% for C max and 18% to 45% for AUC ∞ . The intra-subject variability of esketamine is approximately 15% for C max and 10% for AUC ∞ .
Distribution
The mean steady-state volume of distribution of esketamine administered by the intravenous route is 709 L.
Protein binding of esketamine was approximately 43% to 45%.
The brain-to-plasma ratio of noresketamine is 4- to 6-times lower than that of esketamine.
Elimination
After C max was reached following intranasal administration, the decline in plasma esketamine concentrations was biphasic, with rapid decline for the initial 2 to 4 hours and a mean terminal half-life (t 1/2 ) that ranged from 7 to 12 hours. The mean clearance of esketamine is approximately 89 L/hour following intravenous administration. The elimination of the major metabolite, noresketamine, from plasma is slower than esketamine. The decline of noresketamine plasma concentrations is biphasic, with rapid decline for the initial 4 hours and a mean terminal t 1/2 of approximately 8 hours.
Metabolism
Esketamine is primarily metabolized to noresketamine metabolite via cytochrome P450 (CYP) enzymes CYP2B6 and CYP3A4 and to a lesser extent CYP2C9 and CYP2C19. Noresketamine is metabolized via CYP-dependent pathways and certain subsequent metabolites undergo glucuronidation.
Excretion
Less than 1% of a dose of nasal esketamine is excreted as unchanged drug in urine. Following intravenous or oral administration, esketamine-derived metabolites were primarily recovered in urine (≥ 78% of a radiolabeled dose) and to a lesser extent in feces (≤ 2% of a radiolabeled dose).
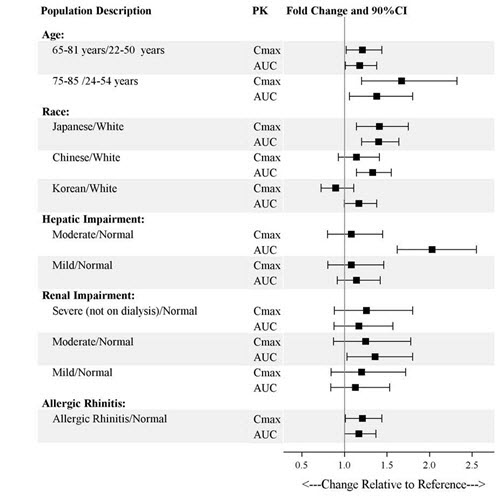
Specific Populations
Exposures of esketamine in specific populations are summarized in Figure 1. No significant differences in the pharmacokinetics of SPRAVATO nasal spray were observed for sex and total body weight (>39 to 170 kg) based on population PK analysis. There is no clinical experience with SPRAVATO nasal spray in patients on renal dialysis or with severe (Child-Pugh class C) hepatic impairment.
Figure 1: Effect of Specific Populations on the Pharmacokinetics of Esketamine
Drug Interaction Studies
The effect of other drugs on the exposures of intranasally administered esketamine are summarized in Figure 2. The effect of SPRAVATO on the exposures of other drugs are summarized in Figure 3. Based on these results, none of the drug-drug interactions are clinically significant.
Figure 2: Effect of Co-administered Drugs on the Pharmacokinetics of Esketamine
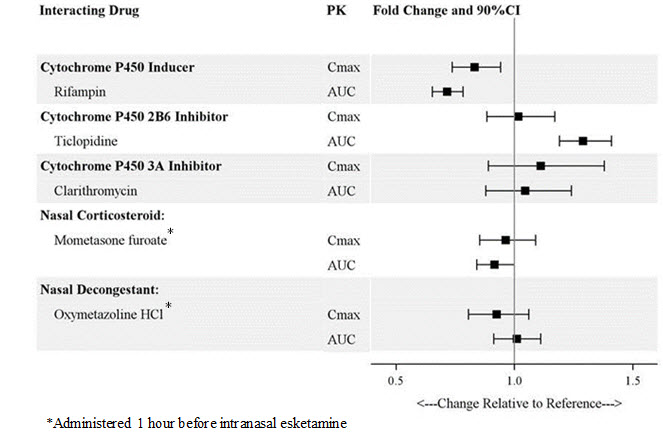
Figure 3: Effect of Esketamine on the Pharmacokinetics of Co-Administered Drugs
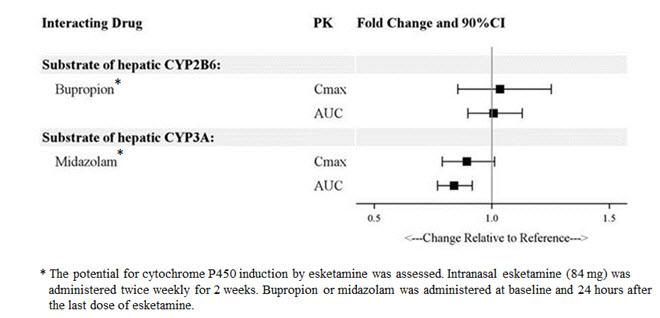
In Vitro Studies
Enzyme Systems : Esketamine has modest induction effects on CYP2B6 and CYP3A4 in human hepatocytes. Esketamine and its major metabolites do not induce CYP1A2. Esketamine and its major circulating metabolites did not show inhibition potential against CYPs and UGTs, except for a weak reversible inhibition of noresketamine on CYP3A4.
Transporter Systems : Esketamine is not a substrate of transporters P-glycoprotein (P-gp; multidrug resistance protein 1), breast cancer resistance protein (BCRP), or organic anion transporter (OATP) 1B1, or OATP1B3. Esketamine and its major circulating metabolites do not inhibit these transporters or multi-drug and toxin extrusion 1 (MATE1) and MATE2-K, or organic cation transporter 2 (OCT2), OAT1, or OAT3.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Once-daily intranasal administration of esketamine at doses equivalent to 4.5, 15, and 45 mg/kg/day (based on a 200-gram rat) did not increase the incidence of tumors in a 2-year rat carcinogenicity study. At the highest dose, the AUC exposure to esketamine was lower than the human exposure (AUC) at the maximum recommended human dose (MRHD) of 84 mg. Once-daily subcutaneous administration of esketamine up to 75 mg/kg/day (reduced to 40 mg/kg/day during week 17) did not increase the incidence of tumors in a 6-month study in transgenic (Tg.rasH2) mice.
Mutagenesis
Esketamine was not mutagenic with or without metabolic activation in the Ames test. Genotoxic effects with esketamine were seen in a screening in vitro micronucleus test in the presence of metabolic activation. However, intravenously-administered esketamine was devoid of genotoxic properties in an in vivo bone marrow micronucleus test in rats and an in vivo Comet assay in rat liver cells.
Impairment of Fertility
Esketamine was administered intranasally to both male and female rats before mating, throughout the mating period, and up to day 7 of gestation at doses equivalent to 4.5, 15, and 45 mg/kg/day (based on a 200-gram rat), which are approximately 0.05, 0.3, and 0.6-times the maximum recommended human dose (MRHD) of 84 mg/day based on mean AUC exposures, respectively. Estrous cycle irregularities were observed at the high dose of 45 mg/kg/day and increased time to mate was observed at doses ≥ 15 mg/kg/day without an overall effect on mating or fertility indices. The No Observed Adverse Effect Level (NOAEL) for mating and fertility is 45 mg/kg/day which is 0.6 times the esketamine exposures at MRHD of 84 mg/day.
Animal Toxicology and/or Pharmacology
Neurotoxicity
In a single-dose neuronal toxicity study where esketamine was administered intranasally to adult female rats, there were no findings of neuronal vacuolation in the brain up to an estimated dose equivalent of 45 mg/kg for a 200-gram rat with a safety margin of 1.8 and 4.5 times the clinical exposures for AUC and C max , respectively, to the MRHD of 84 mg/day. In a second single-dose neurotoxicity study conducted with intranasally administered esketamine to adult female rats, there were no findings of neuronal necrosis up to a dose equivalent of 270 mg/kg for a 200-gram rat which has a safety margin of 18-fold and 23-fold, respectively, to AUC and C max exposures at the MRHD of 84 mg/day. Neuronal vacuolation was not examined in this study.
In a single-dose neuronal toxicity study in adult rats, subcutaneously administered racemic ketamine caused neuronal vacuolation in layer I of the retrosplenial cortex of the brain without neuronal necrosis at a dose of 60 mg/kg. The NOAEL for vacuolation in this study was 15 mg/kg. Estimating 50% of the exposure to be from esketamine, the NOAEL for neuronal vacuolation is 1.6-times and 4.5-times and the NOAEL for neuronal necrosis is 10-times and 16-times exposures, respectively, for AUC and C max to the clinical exposure at the MRHD of 84 mg/day. The relevance of these findings to humans is unknown.
CLINICAL STUDIES
Treatment-Resistant Depression
Short-Term Study
SPRAVATO was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week), Phase 3 study (Study 1; NCT02418585) in adult patients 18 to <65 years old with treatment-resistant depression (TRD). Patients in Study 1 met DSM-5 criteria for major depressive disorder (MDD) and in the current depressive episode, had not responded adequately to at least two different antidepressants of adequate dose and duration. After discontinuing prior antidepressant treatments, patients in Study 1 were randomized to receive twice weekly doses of intranasal SPRAVATO (flexible dose; 56 mg or 84 mg) or intranasal placebo. All patients also received open-label concomitant treatment with a newly initiated daily oral antidepressant (AD) (duloxetine, escitalopram, sertraline, or extended-release venlafaxine as determined by the investigator based on patient's prior treatment history). SPRAVATO could be titrated up to 84 mg starting with the second dose based on investigator discretion.
The demographic and baseline disease characteristics of patients in Study 1 were similar for the SPRAVATO and placebo nasal spray groups. Patients had a median age of 47 years (range 19 to 64 years) and were 62% female, 93% Caucasian, and 5% Black. The newly initiated oral AD was an SSRI in 32% of patients and an SNRI in 68% of patients.
In Study 1, the primary efficacy measure was change from baseline in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score at the end of the 4-week double-blind induction phase. The MADRS is a ten-item, clinician-rated scale used to assess severity of depressive symptoms. Scores on the MADRS range from 0 to 60, with higher scores indicating more severe depression. SPRAVATO plus a newly initiated oral AD demonstrated statistical superiority on the primary efficacy measure compared to placebo nasal spray plus a newly initiated oral AD (see Table 10 ).
| Treatment Group | Number of Patients | Mean Baseline Score (SD) | LS Mean (SE) Change from Baseline to end of Week 4 | LS Mean Difference (95% CI) Difference (SPRAVATO + Oral AD minus Placebo nasal spray + Oral AD) in least-squares mean change from baseline |
|---|---|---|---|---|
| SD=standard deviation; SE=standard error; LS Mean=least-squares mean; CI=confidence interval; AD=antidepressant | ||||
| SPRAVATO (56 mg or 84 mg) + Oral AD SPRAVATO + Oral AD was statistically significantly superior to placebo nasal spray + oral AD | 114 | 37.0 (5.7) | -19.8 (1.3) | -4.0 (-7.3; -0.6) |
| Placebo nasal spray + Oral AD | 109 | 37.3 (5.7) | -15.8 (1.3) | - |
Time Course of Treatment Response
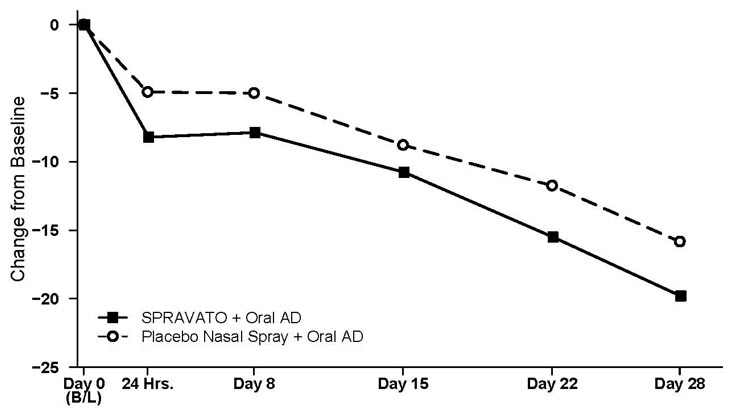
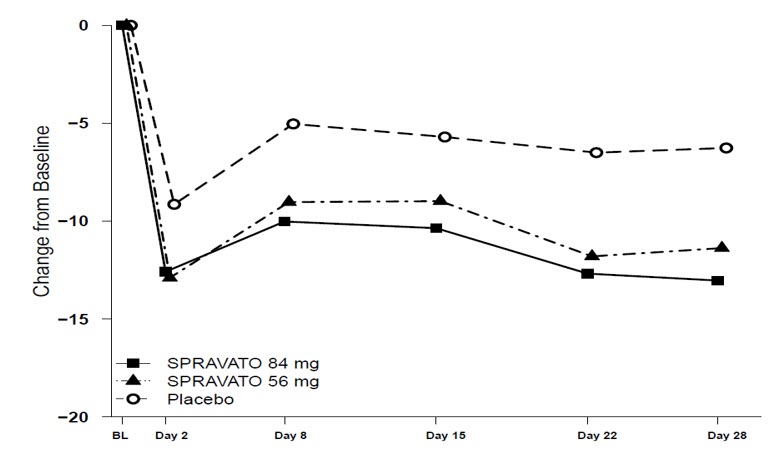
Figure 4 shows the time course of response for the primary efficacy measure (MADRS) in Study 1. Most of SPRAVATO's treatment difference compared to placebo was observed at 24 hours. Between 24 hours and Day 28, both the SPRAVATO and placebo groups continued to improve; the difference between the groups generally remained but did not appear to increase over time through Day 28. At Day 28, 67% of the patients randomized to SPRAVATO were receiving 84 mg twice weekly.
| Figure 4: Least Squares Mean Change from Baseline in MADRS Total Score Over Time in Patients with TRD in Study 1 Note: In this flexible-dose study, dosing was individualized based on efficacy and tolerability. Few subjects (<10%) had reduction in SPRAVATO dosage from 84 mg to 56 mg twice weekly. (Full Analysis Set) |
 |
Treatment-Resistant Depression – Long-term Study
Study 2 (NCT02493868) was a long-term randomized, double-blind, parallel-group, multicenter maintenance-of-effect study in adults 18 to <65 years of age who were known remitters and responders to SPRAVATO. Patients in this study were responders in one of two short-term controlled trials (Study 1 and another 4-week study) or in an open-label direct-enrollment study in which they received flexibly-dosed SPRAVATO (56 mg or 84 mg twice weekly) plus daily oral AD in an initial 4-week phase.
Stable remission was defined as a MADRS total score ≤ 12 for at least 3 of the last 4 weeks. Stable response was defined as a MADRS total score reduction ≥ 50% for the last 2 weeks of optimization and not in remission. After at least 16 initial weeks of treatment with SPRAVATO and an oral AD, stable remitters and stable responders were randomized separately to continue intranasal treatment with SPRAVATO or switch to placebo nasal spray, in both cases with continuation of their oral AD. The primary study endpoint was time to relapse in the stable remitter group. Relapse was defined as a MADRS total score ≥22 for 2 consecutive weeks or hospitalization for worsening depression or any other clinically relevant event indicative of relapse.
The demographic and baseline disease characteristics of the two groups were similar. Patients had a median age of 48 years (range 19 to 64 years) and were 66% female, 90% Caucasian, and 4% Black.
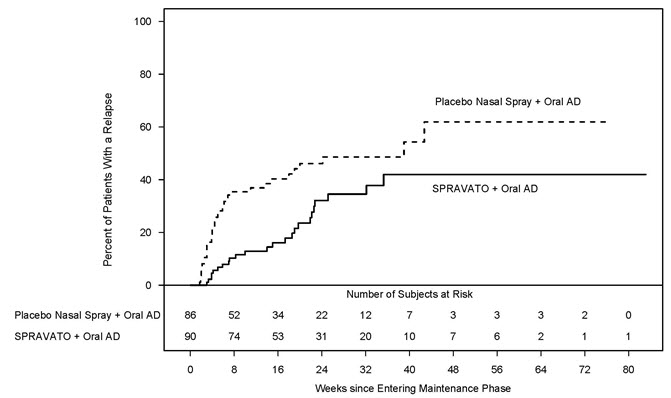
Patients in stable remission who continued treatment with SPRAVATO plus oral AD experienced a statistically significantly longer time to relapse of depressive symptoms than did patients on placebo nasal spray plus an oral AD (see Figure 5 ).
| Figure 5: Time to Relapse in Patients with TRD in Stable Remission in Study 2 Note: The estimated hazard ratio (95% CI) of SPRAVATO + Oral AD relative to Placebo nasal spray + Oral AD based on weighted estimates was 0.49 (95% CI: 0.29, 0.84). However, the hazard ratio did not appear constant throughout the trial. (Full Analysis Set) |
 |
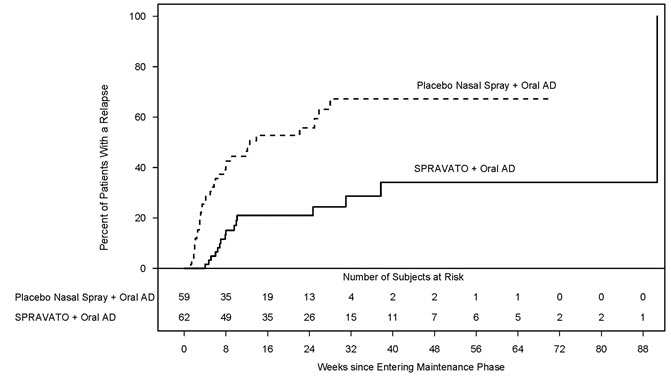
Time to relapse was also significantly delayed in the stable responder population. These patients experienced a statistically significantly longer time to relapse of depressive symptoms than patients on placebo nasal spray plus oral AD (see Figure 6 ).
| Figure 6: Time to Relapse in Patients in Stable Response in Patients with TRD in Study 2 Note: The estimated hazard ratio (95% CI) of SPRAVATO + Oral AD relative to Placebo nasal spray + Oral AD based on Cox proportional hazards model was 0.30 (95% CI: 0.16, 0.55). (Full Analysis Set) |
 |
In Study 2, based on depressive symptomatology, the majority of stable remitters (69%) received every-other-week dosing for the majority of time during the maintenance phase; 23% of stable remitters received weekly dosing. Among stable responders, 34% received every-other-week dosing and 55% received weekly dosing the majority of time during the maintenance phase. Of the patients randomized to SPRAVATO, 39% received the 56 mg dose and 61% received the 84 mg dose.
Monotherapy Study
SPRAVATO was evaluated in a randomized, double-blind, placebo-controlled, multicenter study (Study 3; NCT04599855) in adult patients with treatment resistant depression (TRD) to evaluate the efficacy, safety, and tolerability of SPRAVATO nasal spray, 56 mg and 84 mg, administered as monotherapy. Patients in Study 3 met DSM-5 criteria for major depressive disorder (MDD) and in the current depressive episode, had not responded adequately to at least two different antidepressants of adequate dose and duration. After discontinuing prior antidepressant treatments if applicable, patients in Study 3 were randomized in a 1:1:2 ratio to receive twice weekly doses of intranasal SPRAVATO 56 mg or 84 mg or intranasal placebo for four weeks.
The demographic and baseline disease characteristics of patients in Study 3 were similar for the SPRAVATO and placebo nasal spray groups. Patients had a median age of 46 years (range 19 to 76 years) and were 61% female, 87% Caucasian, and 7% Black.
In Study 3, the primary efficacy measure was change from baseline in the (MADRS) total score at Day 28. SPRAVATO 56 mg and 84 mg monotherapy demonstrated statistical superiority on the primary efficacy measure compared to placebo nasal spray (see Table 11 ).
| Treatment Group | Number of Patients | Mean Baseline Score (SD) | LS Mean (SE) Change from Baseline at Day 28 | LS Mean Difference (95% CI) Difference (SPRAVATO minus Placebo nasal spray) in least-squares mean change from baseline |
|---|---|---|---|---|
| SD=standard deviation; SE=standard error; LS Mean=least-squares mean; CI=confidence interval | ||||
| SPRAVATO (56 mg) SPRAVATO monotherapy is statistically significantly superior to placebo nasal spray | 86 | 37.5 (5.2) | -11.4 (1.2) | -5.1 (-7.9; -2.3) |
| SPRAVATO (84 mg) | 95 | 36.6 (4.5) | -13.0 (1.2) | -6.8 (-9.5; -4.1) |
| Placebo nasal spray | 197 | 37.5 (4.9) | -6.3 (0.8) | - |
In Study 3 the key secondary endpoint was change from baseline to Day 2 (approximately 24 hours) in the MADRS total score. The improvement at Day 2 by SPRAVATO 56 mg and 84 mg monotherapy demonstrated statistical superiority when compared to placebo nasal spray.
Time Course of Treatment Response
Figure 7 shows the time course of response for the primary efficacy measure (MADRS) in Study 3. The effect of SPRAVATO (56 mg and 84 mg) was observed at Day 2 (approximately 24 hours) and remained through Day 28.
Figure 7: Least Squares Mean Change from Baseline in MADRS Total Score Over Time in Patients with TRD in Study 3 (Full Analysis Set)
Depressive Symptoms in Patients with Major Depressive Disorder with Acute Suicidal Ideation or Behavior
SPRAVATO was evaluated in two identical Phase 3 short-term (4-week) randomized, double-blind, multicenter, placebo-controlled studies, Study 4 (NCT03039192) and Study 5 (NCT03097133), in adults with moderate-to-severe MDD (MADRS total score >28) who had active suicidal ideation and intent. In these studies, patients received treatment with SPRAVATO 84 mg or placebo nasal spray twice-weekly for 4 weeks. After the first dose, a one-time dose reduction to SPRAVATO 56 mg was allowed for patients unable to tolerate the 84 mg dose. All patients received comprehensive standard of care treatment, including an initial inpatient psychiatric hospitalization and a newly initiated or optimized oral antidepressant (AD) (AD monotherapy or AD plus augmentation therapy) as determined by the investigator. After completion of the 4-week treatment period with SPRAVATO/placebo, study follow-up continued through Day 90.
The baseline demographic and disease characteristics of patients in Study 4 and Study 5 were similar between the SPRAVATO plus standard of care or placebo nasal spray plus standard of care treatment groups. The median patient age was 40 years (range 18 to 64 years), 61% were female; 73% Caucasian and 6% Black; and 63% of patients had at least one prior suicide attempt. Prior to entering the study, 92% of the patients were receiving antidepressant therapy. During the study, as part of standard of care treatment, 40% of patients received AD monotherapy, 54% of patients received AD plus augmentation therapy, and 6% received both AD monotherapy/AD plus augmentation therapy.
The primary efficacy measure was the change from baseline in the MADRS total score at 24 hours after first dose (Day 2). In Study 4 and Study 5, SPRAVATO plus standard of care demonstrated statistical superiority on the primary efficacy measure compared to placebo nasal spray plus standard of care (see Table 12 ).
| Study No. | Treatment Group SOC treatment included an initial inpatient psychiatric hospitalization and a newly initiated or optimized oral antidepressant (antidepressant monotherapy or antidepressant monotherapy plus augmentation therapy). | Number of Patients | Mean Baseline Score (SD) | LS Mean Change from Baseline to 24 hr Post First Dose (SE) | LS Mean Difference (95% CI) Difference (SPRAVATO + SOC minus placebo nasal spray + SOC) in least-squares mean change from baseline. |
|---|---|---|---|---|---|
| SD=standard deviation; SE=standard error; LS Mean=least-squares mean; CI=confidence interval; SOC=standard of care. | |||||
| Study 4 | SPRAVATO 84 mg + SOC SPRAVATO + SOC were statistically significantly superior to placebo nasal spray + SOC. | 111 | 41.3 (5.87) | -15.9 (1.04) | -3.8 (-6.56; -1.09) |
| Placebo nasal spray + SOC | 112 | 41.0 (6.29) | -12.0 (1.02) | - | |
| Study 5 | SPRAVATO 84 mg + SOC | 113 | 39.4 (5.21) | -16.0 (1.02) | -3.9 (-6.60; -1.11) |
| Placebo nasal spray + SOC | 113 | 39.9 (5.76) | -12.2 (1.05) | - | |
The secondary efficacy measure was the change in Clinical Global Impression of Suicidal Severity - Revised (CGI-SS-r) score at 24 hours after first dose (Day 2). The CGI-SS-r is a one-item, clinician-rated assessment used to rate the current severity of a patient's suicidal ideation and behavior. Scores on the CGI-SS-r range from 0 to 6, with higher scores indicating more severe suicidal ideation and behavior. In Study 4 and Study 5, SPRAVATO plus standard of care did not demonstrate superiority compared to placebo nasal spray plus standard of care in improving CGI-SS-r.
Time Course of Treatment Response
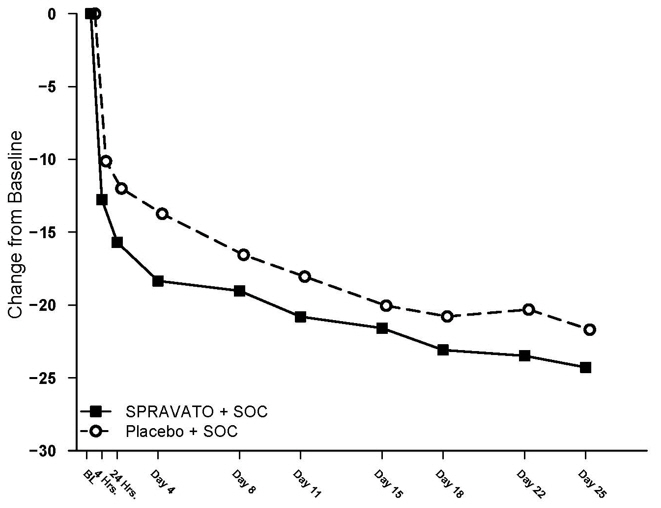
In both Study 4 and Study 5, SPRAVATO's treatment difference compared to placebo was observed starting at 4 hours. Between 4 hours and Day 25, both the SPRAVATO and placebo groups continued to improve; the difference between the groups generally remained but did not appear to increase over time through Day 25. Figure 8 depicts time course of the primary efficacy measure of change in MADRS total score from Study 4.
Figure 8: Least Squares Mean Change from Baseline in MADRS Total Score Over Time in Study 4 Note: In Study 4, after the first dose, a one-time dose reduction to SPRAVATO 56 mg was allowed for patients unable to tolerate the 84 mg dose. Approximately 19% of patients had reduction in SPRAVATO dosage from 84 mg to 56 mg twice weekly. (Full Analysis Set)
Special Safety Studies
Effects on Driving
Two studies were conducted to assess the effects of SPRAVATO on driving skills; one study in adult patients with major depressive disorder (Study 6) and one study in healthy subjects (Study 7). On-road driving performance was assessed by the mean standard deviation of the lateral position (SDLP), a measure of driving impairment.
A single-blind, placebo-controlled study in 25 adult patients with major depressive disorder evaluated the effects of a single 84 mg dose of intranasal SPRAVATO on next day driving and the effect of repeated administration of 84 mg of intranasal SPRAVATO on same-day driving performance (Study 6). For the single dose treatment phase, an ethanol-containing beverage was used as a positive control. The SDLP after administration of single 84 mg dose of SPRAVATO nasal spray was similar to placebo 18 hours post-dose. For the multiple dose treatment phase, the SDLP after repeated administration of 84 mg intranasal SPRAVATO was similar to placebo 6 hours post-dose on Day 11, Day 18, and Day 25.
A randomized, double-blind, cross-over, placebo-controlled study in 23 healthy subjects evaluated the effects of a single 84-mg dose of esketamine nasal spray on driving (Study 7). Mirtazapine (30 mg) was used as a positive control. Driving performance was assessed at 8 hours after SPRAVATO or mirtazapine administration. The SDLP 8 hours after SPRAVATO nasal spray administration was similar to placebo. Two subjects discontinued the driving test after receiving SPRAVATO because of a perceived inability to drive after experiencing post-dose adverse reactions; one subject reported pressure behind the eyes and paresthesia of the hands and feet, the other reported headache with light sensitivity and anxiety.
HOW SUPPLIED/STORAGE AND HANDLING
SPRAVATO ® nasal spray is available as an aqueous solution of esketamine hydrochloride in a stoppered glass vial within a nasal spray device. Each nasal spray device delivers two sprays containing a total of 28 mg of esketamine (supplied as 32.3 mg of esketamine hydrochloride).
SPRAVATO is available in the following presentations:
- 56 mg Dose Kit: Unit-dose carton containing two 28 mg nasal spray devices (56 mg total dose) (NDC 50458-028-02).
- 84 mg Dose Kit: Unit-dose carton containing three 28 mg nasal spray devices (84 mg total dose) (NDC 50458-028-03).
Within each kit, each 28 mg device is individually packaged in a sealed blister (NDC 50458-028-00).
Storage
Store at 20° to 25°C (68° to 77°F); excursions permitted from 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature] .
Disposal
SPRAVATO nasal spray devices must be handled with adequate security, accountability, and proper disposal, per facility procedure for a Schedule III drug product, and per applicable federal, state, and local regulations.
INSTRUCTIONS FOR USE
SPRAVATO ® (SPRAH VAH' TOE) CIII (esketamine) Nasal Spray Device
 |
| 28 mg per device Each device delivers two sprays containing a total of 28 mg of esketamine. |
Important
This device is intended for administration by the patient, under supervision of a healthcare professional . Read this Instructions for Use in full before training and supervising patient.
 Need help?
Need help?
For additional assistance or to share your feedback call 800-JANSSEN (800-526-7736).
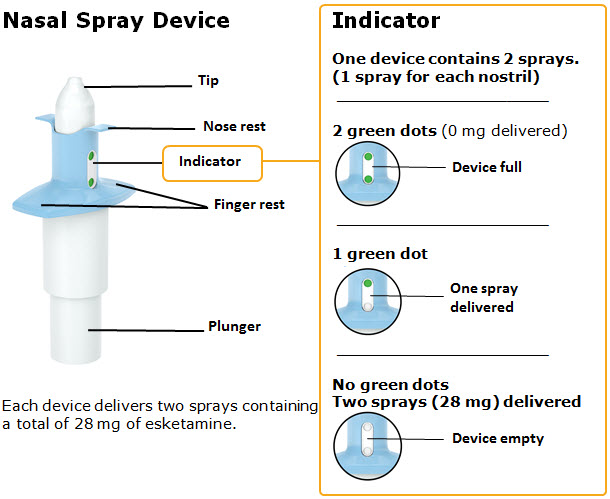

Before first device only:
| Instruct patient to blow nose before first device only . |
| Confirm required number of devices. |


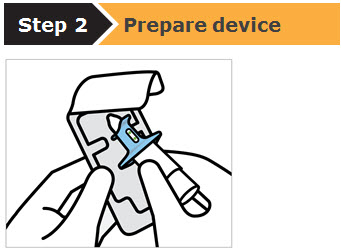
Healthcare professional:
- Check expiration date ('EXP'). If expired, get a new device.
- Peel blister and remove device.
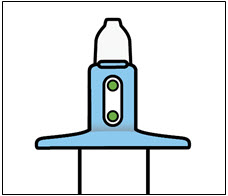
Healthcare professional:
- Do not prime device. This will result in a loss of medication.
- Check that indicator shows 2 green dots . If not, dispose of device and get a new one.
- Hand device to patient.
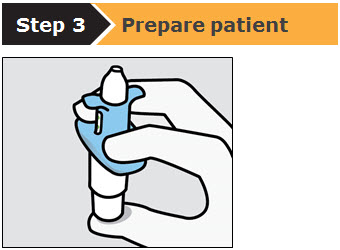
Instruct the patient to:
- Hold device as shown with the thumb gently supporting the plunger.
- Do not press the plunger.

Instruct the patient to:
- Recline head at about 45 degrees during administration to keep medication inside the nose.
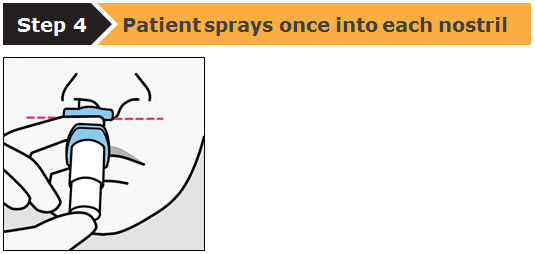
Instruct the patient to:
- Insert tip straight into the first nostril .
- Nose rest should touch the skin between the nostrils .
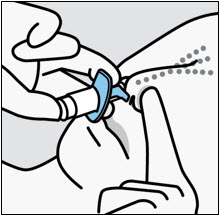
Instruct the patient to:
- Close opposite nostril.
- Breathe in through nose while pushing plunger all the way up until it stops.
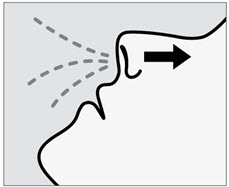
Instruct the patient to:
- Sniff gently after spraying to keep medication inside nose.
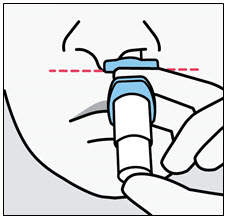
Instruct the patient to:
- Switch hands to insert tip into the second nostril .
- Repeat Step 4 to deliver second spray.
Healthcare professional:
- Take device from patient.
- Check that indicator shows no green dots. If you see a green dot, have patient spray again into the second nostril.
- Check indicator again to confirm device is empty.
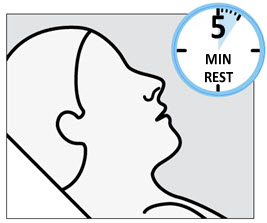
Instruct the patient to:
- Rest in a comfortable position (preferably, semi-reclined) for 5 minutes after each device .
- If liquid drips out, dab nose with a tissue.
 Do not blow nose.
Do not blow nose.
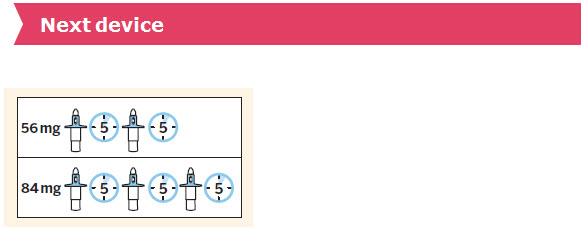
Healthcare professional:
- Repeat Steps 2–5 for the next device.

This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured for: Janssen Pharmaceuticals, Inc. Titusville, NJ 08560
Revised: November 2019
© 2019 Janssen Pharmaceutical Companies
Mechanism of Action
Esketamine, the S-enantiomer of racemic ketamine, is a non-selective, non-competitive antagonist of the N -methyl- D -aspartate (NMDA) receptor, an ionotropic glutamate receptor. The mechanism by which esketamine exerts its antidepressant effect is unknown. The major circulating metabolite of esketamine (noresketamine) demonstrated activity at the same receptor with less affinity.