Get your patient on Vyzulta (Latanoprostene Bunod)
Vyzulta patient education
Patient toolkit
Dosage & administration

Vyzulta prescribing information
INDICATIONS AND USAGE
VYZULTA ® (latanoprostene bunod ophthalmic solution) 0.024% is indicated for the reduction of intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension.
DOSAGE AND ADMINISTRATION
The recommended dosage is one drop in the conjunctival sac of the affected eye(s) once daily in the evening. Do not administer VYZULTA (latanoprostene bunod ophthalmic solution), 0.024% more than once daily since it has been shown that more frequent administration of prostaglandin analogs may lessen the intraocular pressure lowering effect.
If VYZULTA is to be used concomitantly with other topical ophthalmic drug products to lower intraocular pressure, administer each drug product at least five (5) minutes apart.
DOSAGE FORMS AND STRENGTHS
VYZULTA is a topical ophthalmic solution containing latanoprostene bunod, 0.24 mg/mL.
USE IN SPECIFIC POPULATIONS
Pregnancy
Risk Summary
There are no available human data for the use of VYZULTA during pregnancy to inform any drug associated risks.
Latanoprostene bunod has caused miscarriages, abortion, and fetal harm in rabbits. Latanoprostene bunod was shown to be abortifacient and teratogenic when administered intravenously (IV) to pregnant rabbits at exposures ≥ 0.28 times the clinical dose. Doses ≥ 20 mcg/kg/day (23 times the clinical dose) produced 100% embryofetal lethality. Structural abnormalities observed in rabbit fetuses included anomalies of the great vessels and aortic arch vessels, domed head, sternebral and vertebral skeletal anomalies, limb hyperextension and malrotation, abdominal distension and edema. Latanoprostene bunod was not teratogenic in the rat when administered IV at 150 mcg/kg/day (87 times the clinical dose) [see Data] .
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk in the U.S. general population of major birth defects is 2 to 4%, and of miscarriage is 15 to 20%, of clinically recognized pregnancies.
Data
Animal Data
Embryofetal studies were conducted in pregnant rabbits administered latanoprostene bunod daily by intravenous injection on gestation days 7 through 19, to target the period of organogenesis. The doses administered ranged from 0.24 to 80 mcg/kg/day. Abortion occurred at doses ≥ 0.24 mcg/kg/day latanoprostene bunod (0.28 times the clinical dose, on a body surface area basis, assuming 100% absorption). Embryofetal lethality (resorption) was increased in latanoprostene bunod treatment groups, as evidenced by increases in early resorptions at doses ≥ 0.24 mcg/kg/day and late resorptions at doses ≥ 6 mcg/kg/day (approximately 7 times the clinical dose). No fetuses survived in any rabbit pregnancy at doses of 20 mcg/kg/day (23 times the clinical dose) or greater. Latanoprostene bunod produced structural abnormalities at doses ≥ 0.24 mcg/kg/day (0.28 times the clinical dose). Malformations included anomalies of sternum, coarctation of the aorta with pulmonary trunk dilation, retroesophageal subclavian artery with absent brachiocephalic artery, domed head, forepaw hyperextension and hindlimb malrotation, abdominal distention/edema, and missing/fused caudal vertebrae.
An embryofetal study was conducted in pregnant rats administered latanoprostene bunod daily by intravenous injection on gestation days 7 through 17, to target the period of organogenesis. The doses administered ranged from 150 to 1500 mcg/kg/day. Maternal toxicity was produced at 1500 mcg/kg/day (870 times the clinical dose, on a body surface area basis, assuming 100% absorption), as evidenced by reduced maternal weight gain. Embryofetal lethality (resorption and fetal death) and structural anomalies were produced at doses ≥ 300 mcg/kg/day (174 times the clinical dose). Malformations included anomalies of the sternum, domed head, forepaw hyperextension and hindlimb malrotation, vertebral anomalies and delayed ossification of distal limb bones. A no observed adverse effect level (NOAEL) was established at 150 mcg/kg/day (87 times the clinical dose) in this study.
Lactation
Risk Summary
There are no data on the presence of VYZULTA in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for VYZULTA, and any potential adverse effects on the breastfed infant from VYZULTA.
Pediatric Use
Use in pediatric patients aged 16 years and younger is not recommended because of potential safety concerns related to increased pigmentation following long-term chronic use.
Geriatric Use
No overall clinical differences in safety or effectiveness have been observed between elderly and other adult patients.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
- Pigmentation: Increased pigmentation of the iris and periorbital tissue (eyelid) can occur. Iris pigmentation is likely to be permanent. (5.1 )
- Eyelash changes: Gradual changes to eyelashes including increased length, increased thickness and number of eyelashes. Usually reversible upon discontinuation of treatment. (5.2 )
Pigmentation
VYZULTA (latanoprostene bunod ophthalmic solution), 0.024% may cause changes to pigmented tissues. The most frequently reported changes with prostaglandin analogs have been increased pigmentation of the iris and periorbital tissue (eyelid).
Pigmentation is expected to increase as long as latanoprostene bunod ophthalmic solution is administered. The pigmentation change is due to increased melanin content in the melanocytes rather than to an increase in the number of melanocytes. After discontinuation of VYZULTA, pigmentation of the iris is likely to be permanent, while pigmentation of the periorbital tissue and eyelash changes are likely to be reversible in most patients. Patients who receive prostaglandin analogs, including VYZULTA, should be informed of the possibility of increased pigmentation, including permanent changes. The long-term effects of increased pigmentation are not known.
Iris color change may not be noticeable for several months to years. Typically, the brown pigmentation around the pupil spreads concentrically towards the periphery of the iris and the entire iris or parts of the iris become more brownish. Neither nevi nor freckles of the iris appear to be affected by treatment. While treatment with VYZULTA (latanoprostene bunod ophthalmic solution), 0.024% can be continued in patients who develop noticeably increased iris pigmentation, these patients should be examined regularly [see Patient Counseling Information (17) ].
Eyelash Changes
VYZULTA may gradually change eyelashes and vellus hair in the treated eye. These changes include increased length, thickness, and the number of lashes or hairs. Eyelash changes are usually reversible upon discontinuation of treatment.
Intraocular Inflammation
VYZULTA should be used with caution in patients with a history of intraocular inflammation (iritis/uveitis) and should generally not be used in patients with active intraocular inflammation as it may exacerbate this condition.
Macular Edema
Macular edema, including cystoid macular edema, has been reported during treatment with prostaglandin analogs. VYZULTA should be used with caution in aphakic patients, in pseudophakic patients with a torn posterior lens capsule, or in patients with known risk factors for macular edema.
Bacterial Keratitis
There have been reports of bacterial keratitis associated with the use of multiple-dose containers of topical ophthalmic products. These containers had been inadvertently contaminated by patients who, in most cases, had a concurrent corneal disease or a disruption of the ocular epithelial surface.
Use with Contact Lens
Contact lenses should be removed prior to the administration of VYZULTA because this product contains benzalkonium chloride. Lenses may be reinserted 15 minutes after administration.
ADVERSE REACTIONS
The following adverse reactions are described elsewhere in the labeling:
- Pigmentation [see Warnings and Precautions (5.1) ]
- Eyelash Changes [see Warnings and Precautions (5.2) ]
- Intraocular Inflammation [see Warnings and Precautions (5.3) ]
- Macular Edema [see Warnings and Precautions (5.4) ]
- Bacterial Keratitis [see Warnings and Precautions (5.5) ]
- Use with Contact Lens [see Warnings and Precautions (5.6) ]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
VYZULTA was evaluated in 811 patients in 2 controlled clinical trials of up to 12 months duration. The most common ocular adverse reactions observed in patients treated with latanoprostene bunod were: conjunctival hyperemia (6%), eye irritation (4%), eye pain (3%), and instillation site pain (2%). Approximately 0.6% of patients discontinued therapy due to ocular adverse reactions including ocular hyperemia, conjunctival irritation, eye irritation, eye pain, conjunctival edema, vision blurred, punctate keratitis and foreign body sensation.
DESCRIPTION
VYZULTA ® (latanoprostene bunod ophthalmic solution), 0.024% is a prostaglandin analog formulated as a sterile topical ophthalmic solution. VYZULTA contains the active ingredient latanoprostene bunod 0.24 mg/mL, the preservative benzalkonium chloride 0.2 mg/mL, and the following inactive ingredients: polysorbate 80, glycerin, EDTA, and water. The formulation is buffered to pH 5.5 with citric acid/sodium citrate.
Its chemical name is 4-(Nitrooxy)butyl (5Z)-7-{(1R,2R,3R,5S)-3,5-dihydroxy-2-[(3R)-3-hydroxy-5-phenylpentyl]cyclopentyl}hept-5-enoate. Its molecular formula is C 27 H 41 NO 8 . Molecular weight: 507.62.
Its chemical structure is:
Figure 1
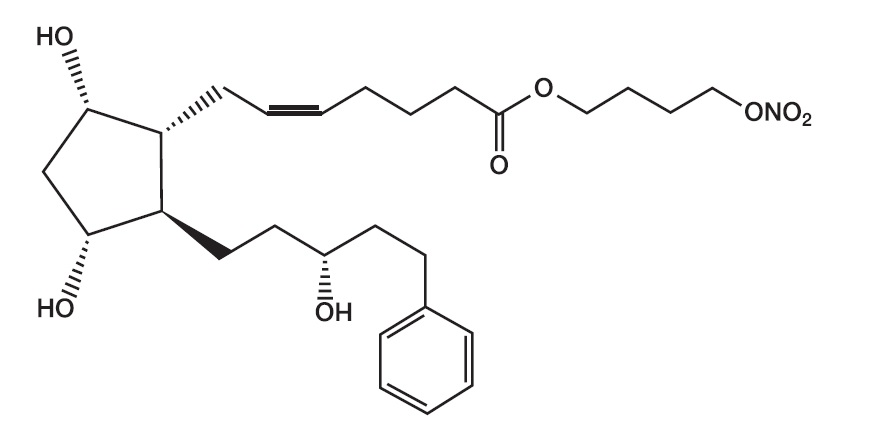
Latanoprostene bunod is a colorless to yellow oil.
CLINICAL PHARMACOLOGY
Mechanism of Action
Latanoprostene bunod is thought to lower intraocular pressure by increasing outflow of aqueous humor through both the trabecular meshwork and uveoscleral routes. Intraocular pressure is a major modifiable risk factor for glaucoma progression. Reduction of intraocular pressure reduces risk of glaucomatous visual field loss.
Pharmacodynamics
Reduction of the intraocular pressure starts approximately 1 to 3 hours after the first administration with the maximum effect reached after 11-13 hours in eyes with elevated intraocular pressure.
Pharmacokinetics
Absorption
The systemic exposure of latanoprostene bunod and its metabolites latanoprost acid and butanediol mononitrate were evaluated in one study with 22 healthy subjects after topical ocular administration of VYZULTA 0.024% once daily (one drop bilaterally in the morning) for 28 days. There were no quantifiable plasma concentrations of latanoprostene bunod (lower limit of quantitation, LLOQ, of 10.0 pg/mL) or butanediol mononitrate (LLOQ of 200 pg/mL) post-dose on Day 1 and Day 28. The mean maximal plasma concentrations (Cmax) of latanoprost acid (LLOQ of 30 pg/mL) were 59.1 pg/mL and 51.1 pg/mL on Day 1 and Day 28, respectively. The mean time of maximal plasma concentration (Tmax) for latanoprost acid was approximately 5 minutes post-administration on both Day 1 and Day 28.
Distribution
There were no ocular distribution studies performed in humans.
Metabolism
After topical ocular administration, latanoprostene bunod is rapidly metabolized in the eye to latanoprost acid (active moiety), an F2α prostaglandin analog, and butanediol mononitrate. After latanoprost acid reaches the systemic circulation, it is primarily metabolized by the liver to the 1,2-dinor and 1,2,3,4-tetranor metabolites via fatty acid β-oxidation.
Butanediol mononitrate is metabolized to 1,4-butanediol and nitric oxide. The metabolite 1,4-butanediol is further oxidized to succinic acid and enters the tricarboxylic acid (TCA) cycle.
Elimination
The elimination of latanoprost acid from human plasma is rapid as latanoprost acid plasma concentration dropped below the LLOQ (30 pg/mL) in the majority of subjects by 15 minutes following ocular administration of VYZULTA 0.024% in humans.
NONCLINICAL TOXICOLOGY
Carcinogenesis, Mutagenesis, Impairment of Fertility
Latanoprostene bunod was not mutagenic in bacteria and did not induce micronuclei formation in the in vivo rat bone marrow micronucleus assay. Chromosomal aberrations were observed in vitro with human lymphocytes in the absence of metabolic activation.
Latanoprostene bunod has not been tested for carcinogenic activity in long-term animal studies. Latanoprost acid is a main metabolite of latanoprostene bunod. Exposure of rats and mice to latanoprost acid, resulting from oral dosing with latanoprost in lifetime rodent bioassays, was not carcinogenic.
Fertility studies have not been conducted with latanoprostene bunod. The potential to impact fertility can be partially characterized by exposure to latanoprost acid, a common metabolite of both latanoprostene bunod and latanoprost. Latanoprost acid has not been found to have any effect on male or female fertility in animal studies.
Animal Toxicology and/or Pharmacology
A 9-month toxicology study administered topical ocular doses of latanoprostene bunod to one eye of cynomolgus monkeys: control (vehicle only), one drop of 0.024% bid, one drop of 0.04% bid and two drops of 0.04% per dose, bid. The systemic exposures are equivalent to 4.2-fold, 7.9-fold, and 13.5-fold the clinical dose, respectively, on a body surface area basis (assuming 100% absorption). Microscopic evaluation of the lungs after 9 months observed pleural/subpleural chronic fibrosis/inflammation in the 0.04% dose male groups, with increasing incidence and severity compared to controls. Lung toxicity was not observed at the 0.024% dose.
CLINICAL STUDIES
In clinical studies up to 12 months duration, patients with open-angle glaucoma or ocular hypertension with average baseline intraocular pressures (IOPs) of 26.7 mmHg, the IOP-lowering effect of VYZULTA (latanoprostene bunod ophthalmic solution) 0.024% once daily (in the evening) was up to 7 to 9 mmHg.
HOW SUPPLIED/STORAGE AND HANDLING
VYZULTA ® (latanoprostene bunod ophthalmic solution), 0.024% is supplied in low density polyethylene bottles with dropper tips and turquoise caps in the following sizes:
2.5 mL fill in a 4 mL white container - NDC 24208-504-02
5 mL fill in a 7.5 mL natural container - NDC 24208-504-05
Storage: Unopened bottle should be stored refrigerated at 2°C to 8°C (36°F to 46°F). Once a bottle is opened it may be stored at 2°C to 25°C (36°F to 77°F) for 8 weeks.
During shipment, bottles may be maintained at temperatures up to 40°C (104°F) for a period not exceeding 14 days.
Protect from light. Protect from freezing.
Mechanism of Action
Latanoprostene bunod is thought to lower intraocular pressure by increasing outflow of aqueous humor through both the trabecular meshwork and uveoscleral routes. Intraocular pressure is a major modifiable risk factor for glaucoma progression. Reduction of intraocular pressure reduces risk of glaucomatous visual field loss.